Hemofili – orsaker, typer, symtom och behandling
Hemofili är en ärftlig blödningsrubbning där koagulationen är nedsatt. Den här artikeln beskriver orsaker, genetik, typer, symtom, diagnostik, behandling och aktuella utvecklingsområden.
Hemofili är en ärftlig störning i blodets förmåga att koagulera som gör att blödningar varar längre än hos personer med normal koagulation. Sjukdomen uppstår när ett eller flera proteiner som behövs i koagulationskaskaden saknas eller är defekta. Det innebär inte nödvändigtvis att mer blod förloras per minut, utan att blödningen inte upphör lika snabbt som normalt. För grundläggande information om blödningens natur se blödning och för mekanismerna i blodets levnadscykler och koagulation läs koagulation.
Bildgalleri
3 Bilder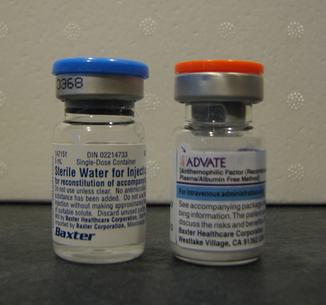

Orsaker och genetik
De vanligaste formerna av hemofili orsakas av mutationer i gener som kodar för koagulationsfaktorer. Hemofili A beror på brist eller fel i faktor VIII och hemofili B på brist i faktor IX. Dessa gener sitter ofta på X-kromosomen, vilket är en förklaring till att män drabbas betydligt oftare än kvinnor. Eftersom män normalt har bara en X-kromosom kan en skadad gen där ge sjukdom, medan kvinnor oftast har en frisk kopia på den andra X-kromosomen som kan maskera en defekt. Läs mer om gener och kromosomer via gener, X-kromosomen och Y-kromosomen. Begreppet könsbunden ärftlighet förklaras i mer detalj hos könsbunden genetik.
Ungefär en tredjedel av fallen uppstår genom en ny mutation i familjen utan tidigare känd ärftlighet. Arvet överförs ofta från en bärare (vanligen en kvinna som inte är sjuk men bär på mutationen) till barnet; därför är information och rådgivning viktiga delar av vården (rådgivningstjänster).
Typer
- Hemofili A – vanligast; orsakad av brist på faktor VIII. Förekomsten anges ofta som cirka 1 av 5 000–10 000 födda pojkar beroende på källa.
- Hemofili B – mindre vanligt; orsakad av brist på faktor IX. Förekomstuppskattningar ligger i storleksordningen 1 av 20 000–30 000 födda pojkar.
- Hemofili C – orsakas av brist på faktor XI och har ett annat ärftlighetsmönster; den är betydligt mer sällsynt i många populationer och kan uppträda i vissa grupper i högre frekvens.
Symtom och komplikationer
Tidiga tecken kan vara onormalt stora blåmärken (blåmärke) efter mindre slag, långdragna blödningar efter skador eller operationer, samt spontana blödningar i leder och muskler. Upprepade blödningar i leder kan med tiden leda till kronisk smärta och ledskador. Inre blödningar, till exempel i bukhålan eller i hjärnan, är allvarliga tillstånd som kräver omedelbart omhändertagande och kan bli livshotande (risk och allvar).
Diagnos
Diagnos baseras på blodprov som mäter aktivitet och nivå av specifika koagulationsfaktorer, kompletterat med anamnes och familjehistoria. Genetisk diagnostik kan bekräfta vilken mutation som förekommer och är viktig för rådgivning om arvsgång och för planering av familjebildning.
Behandling och vård
Standardbehandling är substitutionsbehandling där saknade koagulationsfaktorer ges som koncentrat antingen vid blödningstillfällen eller profylaktiskt för att förebygga blödningar. Moderna preparat kan vara rekombinant framställda och det finns långtidsverkande formuleringar samt andra biologiska terapier. Patientutbildning, individuella vårdplaner och tillgång till specialistvård är centrala. Vid akut blödning kan plasma eller faktorpreparat behövas snabbt; historiskt användes ofta blodtransfusioner. För praktiska råd och patientinformation hänvisas till auktoritativa källor som patientinformation och stöd via lokala rådgivningstjänster.
Förebyggande, egenvård och särskilda situationer
Profylax minskar risk för ledskador och förbättrar livskvaliteten. Personer med hemofili bör ha en individuell behandlingsplan, bära medicinsk ID och informera vårdpersonal vid operationer eller tandvård. Gravida bärare och födande kvinnor behöver särskild rådgivning och planering. Vaccinationer och förebyggande åtgärder mot hepatit och andra infektioner är viktiga då vissa äldre blodprodukter tidigare inneburit smittrisk.
Forskning och framtid
Forskningen inriktas på genteknik, långtidsverkande faktorpreparat, immunmodulerande behandlingar för att hantera antikroppar mot infunderade faktorer, samt nya substanser som kan minska blödningsbenägenheten. Målet är att ytterligare minska komplikationer och möjliggöra ett mer normalt vardagsliv för dem som lever med sjukdomen.
Ytterligare information
För en översikt och vidare läsning om terminologi och relaterade begrepp finns ordlistor och faktablad via ordlistan. För lokala resurser och specialistkliniker rekommenderas kontakt med nationella patientorganisationer och vårdgivare (patientinformation).

Relaterade sidor
Frågor och svar
F: Vad är blödarsjuka?
S: Hemofili är en blodsjukdom som innebär att blödningen inte upphör. Blödningen uppstår eftersom blodet inte koagulerar, på grund av brist på proteiner i blodet som gör skorpor och blodproppar.
F: Hur behandlas blödarsjuka?
S: Blödarsjuka kan behandlas genom att man får en blodgivning från någon som inte har blödarsjuka, eftersom deras blod innehåller koagulationsproteiner som tillfälligt kan göra en normal skorv.
F: Hur vanligt är blödarsjuka?
S: Hemofili A förekommer hos cirka 1 av 5 000-10 000 födda män och hemofili B förekommer hos cirka 1 av 20 000-34 000 födda män.
F: Finns det något botemedel mot denna sjukdom?
S: Det finns inget botemedel mot denna sjukdom, men det finns olika behandlingar tillgängliga runt om i världen.
F: Vad orsakar hemofili?
S: Hemofili drabbar vanligen män och den överförs från mor till barn genom generna. 30 % av de som drabbas av hemofili A och B är den första personen i sin familj som har hemofili, vilket är resultatet av en oväntad mutation (detta innebär att det sker en oväntad förändring i kroppen). Det drabbar vanligtvis män på grund av genetiska defekter på X-kromosomen eftersom de bara har en X-kromosom medan kvinnor har två X-kromosomer så en recessiv gen kan maskeras av en normal gen på den andra X-kromosomen. Defekter av denna typ kallas "könsbundna" inom genetiken.
F: Finns det olika typer av hemofila?
S: Ja, det finns tre typer av hemofilier - hemofilier A (ingen koagulationsförmåga), hemofilier B (otillräcklig koagulationsförmåga) och hemofilier C (orsakade av två recessiva gener).
Relaterade artiklar
Författare
AlegsaOnline.com Hemofili – orsaker, typer, symtom och behandling Leandro Alegsa
URL: https://sv.alegsaonline.com/art/41733
Källor
- etymonline.com : "Online Etymology Dictionary"
- hemophilia.org : "Hemophilia B"