Klinisk prövning – definition, syfte och vanliga studiedesigner
Lär dig vad en klinisk prövning är, dess syfte och vanliga studiedesigner — från placebo till randomisering, för patienter och frivilliga.
En klinisk prövning är ett av de steg som krävs för att införa ett nytt läkemedel eller en ny behandling. Beroende på läkemedlet deltar antingen frivilliga eller patienter som lider av en sjukdom eller ett tillstånd i studien. I allmänhet får ett antal av de personer som deltar det riktiga läkemedlet, eller en bra behandling, och resten av personerna får en behandling eller ett läkemedel som inte har någon effekt, så kallad placebo. Dessa tester är statistiska tester.
Exempel på studiedesigns:
Bildgalleri
6 Bilder




Vad är syftet med en klinisk prövning?
- Säkerhet: Bedöma om läkemedlet eller behandlingen är säker att använda och identifiera biverkningar.
- Effekt: Avgöra om behandlingen har avsedd effekt på sjukdom eller symtom.
- Dosering: Fastställa optimal dos och administrationssätt.
- Jämförelse: Jämföra mot befintlig standardbehandling eller placebo för att visa fördelar eller likvärdighet.
- Regulatoriskt underlag: Ge data som krävs för godkännande av läkemedel och behandlingar.
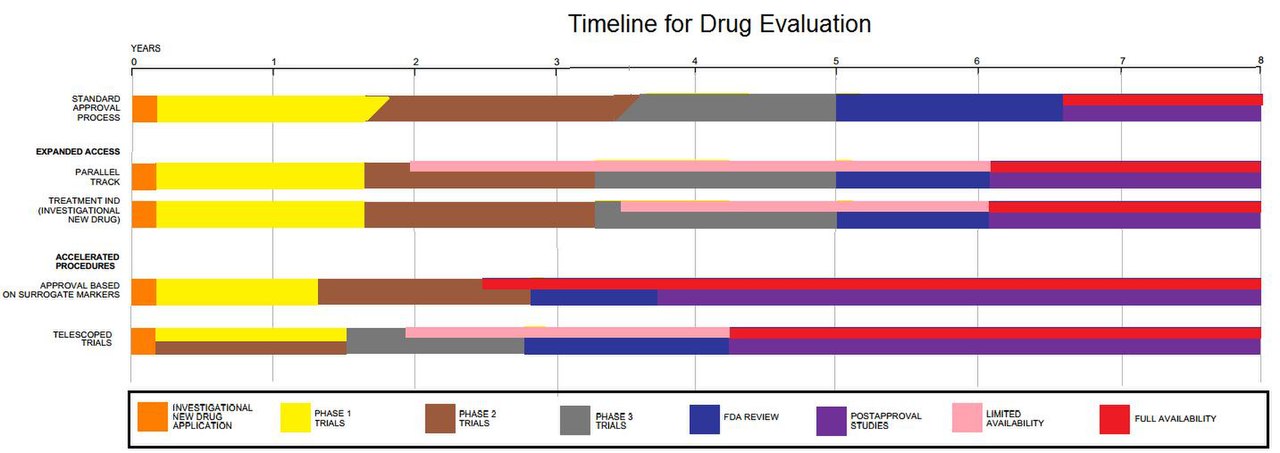
Faser i läkemedelsprövningar
- Fas I: Mindre studier (oftast friska frivilliga) som undersöker säkerhet, tolerans och farmakokinetik (hur kroppen tar upp, fördelar och gör sig av med ett läkemedel).
- Fas II: Mindre till medelstora studier på patienter för att undersöka effekt, dosrespons och fortsatta säkerhetsdata.
- Fas III: Större, ofta randomiserade studier för att bekräfta effekt och säkerhet jämfört med standardbehandling eller placebo. Dessa data används ofta vid registrering hos myndigheter.
- Fas IV: Efter godkännande: uppföljningsstudier för att upptäcka sällsynta biverkningar och studera långtidseffekter i verklig klinisk praxis.
Vanliga studiedesigner
- Randomiserat kontrollerat försök (RCT): Deltagarna slumpas till olika behandlingsgrupper. Ofta dubbelblint (varken deltagare eller forskare vet vilken behandling som ges) för att minska bias.
- Placebokontrollerat: En grupp får placebo. Används när det är etiskt acceptabelt att inte ge aktiv behandling.
- Aktiv kontroll (komparator): Ny behandling jämförs mot befintlig standardbehandling istället för placebo.
- Cross-over: Deltagarna får i tur och ordning båda behandlingarna (med washout-period mellan). Kräver att sjukdomen och behandlingen är lämpliga för denna design.
- Open-label: Deltagare och forskare vet vilken behandling som ges. Används ofta när blindning är svår eller omöjlig.
- Enarmsstudie (single-arm): Alla deltagare får samma behandling — vanligt i tidiga faser eller vid sällsynta sjukdomar.
- Non-inferiority och equivalence: Studiedesigner som visar att en ny behandling inte är sämre än eller är likvärdig med en befintlig behandling.
- Cluster-randomiserade försök: Hela grupper (t.ex. kliniker, skolor) randomiseras snarare än individer.
- Adaptive design: Studie där vissa aspekter (t.ex. dosval, randomiseringsfördelning) kan ändras under pågående prövning utifrån fördefinierade regler och data.
- Pragmatisk prövning: Genomförs i verklig klinisk praxis för att visa effektivitet i vardagliga förhållanden.
Hur en prövning genomförs — steg för steg
- Protokoll: En detaljerad plan beskriver mål, kriterier för att inkludera/exkludera deltagare, behandlingsscheman, endpoints och statistiska metoder.
- Etiskt godkännande: Prövningen måste godkännas av en etikprövningsnämnd och ibland av läkemedelsmyndigheter innan start.
- Informed consent (informerat samtycke): Deltagarna får skriftlig information om prövningen, möjliga risker och förmåner och lämnar frivilligt sitt samtycke.
- Randomisering och blindning: Randomisering minskar urvalsbias; blindning minskar påverkan av förväntningar på resultatet.
- Datainsamling och säkerhetsövervakning: Biverkningar rapporteras och övervakas fortlöpande, ofta av en oberoende Data Safety Monitoring Board (DSMB).
- Analys: Fördefinierade statistiska analyser avgör om målen uppnåtts (t.ex. intention-to-treat vs per-protocol).
- Rapportering och registrering: Resultat publiceras och prövningen registreras i kliniska register för transparens.
Etik, säkerhet och reglering
- Alla kliniska prövningar måste följa principerna i God klinisk praxis (GCP) och nationella lagar.
- Användning av placebo är föremål för etisk prövning — placebo får inte användas när det innebär onödig risk eller utsätter patienter för nedsatt vård.
- Monitorering och snabb rapportering av allvarliga biverkningar är obligatoriskt.
- Många prövningar finansieras av läkemedelsföretag, myndigheter eller akademiska institutioner; oberoende granskning och transparens kring intressekonflikter är viktigt.
Vad deltagare bör veta
- Deltagande är frivilligt och kan avbrytas när som helst utan att normalt vårdutbud påverkas.
- Deltagare får information om möjliga biverkningar, praktiska aspekter (besök, tester) och eventuella ersättningar.
- Kontaktuppgifter till ansvarig prövare och etisk nämnd ska finnas tillgängliga.
- Hälsoutfall kan vara positiva, neutrala eller negativa — inte alla prövningar leder till godkännande av ny behandling.
Fördelar och begränsningar
- Fördelar: Ger vetenskapligt underlag för säkerhet och effekt, kan leda till förbättrad vård och nya behandlingsmöjligheter.
- Begränsningar: Kostsamma och tidskrävande; resultat kan ibland inte generaliseras till alla patientgrupper; tidiga studier kan ha liten storlek och osäkerhet.
Genom att förstå olika studiedesigner och prövningsfaser kan patienter, vårdgivare och allmänheten bättre bedöma vad ett kliniskt prövningsresultat innebär och vilka krav som ställs innan nya läkemedel eller behandlingar blir tillgängliga i vården.
Historia
Kliniska prövningar introducerades för första gången i Avicennas The Canon of Medicine år 1025 e.Kr., där han fastställde regler för experimentell användning och testning av läkemedel. Han skrev en exakt vägledning för praktiska experiment i processen att upptäcka och bevisa effektiviteten hos medicinska läkemedel och substanser. Han fastställde följande regler och principer för att testa effektiviteten hos nya läkemedel och mediciner, som fortfarande utgör grunden för moderna kliniska prövningar:
- Läkemedlet ska vara fritt från alla ovidkommande oavsiktliga egenskaper.
- Den måste användas för en enkel sjukdom, inte en sammansatt sjukdom.
- Läkemedlet måste testas på två olika typer av sjukdomar, eftersom ett läkemedel ibland botar en sjukdom med hjälp av sina väsentliga egenskaper och en annan med hjälp av sina tillfälliga egenskaper.
- Läkemedlets kvalitet måste motsvara sjukdomens styrka. Det finns till exempel vissa läkemedel vars värme är mindre än vissa sjukdomars kyla, så att de inte skulle ha någon effekt på dem.
- Handlingstidpunkten måste iakttas så att väsen och tillfällighet inte förväxlas.
- Man måste se att läkemedlets effekt uppstår ständigt eller i många fall, för om detta inte sker är det en oavsiktlig effekt.
- Försöken måste göras med människokroppen, för att testa ett läkemedel på ett lejon eller en häst kan inte bevisa något om dess effekt på människan.
En av de mest berömda kliniska prövningarna var James Linds demonstration 1747 av att citrusfrukter botar skörbjugg. Han jämförde effekterna av olika sura ämnen, från vinäger till cider, på grupper av drabbade sjömän och fann att den grupp som fick apelsiner och citroner i stort sett hade återhämtat sig från skörbjugg efter sex dagar.
Frederick Akbar Mahomed (död 1884), som arbetade vid Guy's Hospital i London, bidrog i hög grad till den kliniska prövningsprocessen under sina detaljerade kliniska studier, där "han skiljde kronisk nefrit med sekundär hypertoni från vad vi nu kallar essentiell hypertoni". Han grundade också "Collective Investigation Record for the British Medical Association; denna organisation samlade in data från läkare som praktiserade utanför sjukhusmiljön och var föregångare till moderna kliniska prövningar i samarbete".
Frågor och svar
F: Vad är en klinisk prövning?
S: En klinisk prövning är ett viktigt förfarande för att testa nya läkemedel eller terapier innan de släpps till allmänheten.
F: Vem deltar i en klinisk prövning?
S: Frivilliga eller patienter med en sjukdom deltar i en klinisk prövning.
F: Vilka typer av behandlingar får deltagarna i en klinisk prövning?
S: Antingen får deltagarna det läkemedel eller den behandling som testas, eller så får de placebo som inte har någon effekt.
F: Hur genomförs kliniska prövningar?
S: Kliniska prövningar är statistiska tester och innefattar en studiedesign.
F: Varför är kliniska prövningar viktiga?
S: Kliniska prövningar är viktiga för att fastställa effektiviteten och säkerheten hos nya läkemedel eller behandlingar av sjukdomar.
F: Vem har nytta av kliniska prövningar?
S: Patienter som har nytta av nya behandlingar och vårdpersonal som använder dessa behandlingar har nytta av kliniska prövningar.
F: Kan kliniska prövningar avgöra om en ny behandling eller ett nytt läkemedel är säkert?
S: Ja, kliniska prövningar bidrar till att fastställa säkerheten hos nya behandlingar innan de släpps ut till allmänheten.
Relaterade artiklar
Författare
AlegsaOnline.com Klinisk prövning – definition, syfte och vanliga studiedesigner Leandro Alegsa
URL: https://sv.alegsaonline.com/art/21014
Källor
- bruzelius.info : "James Lind: A Treatise of the Scurvy (1754)"