Thalassemi – ärftlig blodsjukdom: symtom, orsaker och behandling
Thalassemi — allt om en ärftlig blodsjukdom: symtom, orsaker, diagnos och behandlingar, komplikationer och möjlig bot med benmärgstransplantation.
Thalassemi (eller thalassemi) är en genetisk sjukdom i blodet som har sitt ursprung i Medelhavsområdet.
Denna sjukdom orsakas av att de röda blodkropparna försvagas och förstörs. Orsaken är muterade gener som påverkar hur kroppen tillverkar hemoglobin. Hemoglobin är det protein i röda blodkroppar som transporterar syre. Personer med thalassemi producerar mindre hemoglobin och färre cirkulerande röda blodkroppar än normalt, vilket resulterar i mild eller svår anemi.
Thalassemi kan orsaka betydande komplikationer, bland annat lunginflammation, järnöverbelastning, benmissbildningar och kardiovaskulära sjukdomar. Denna ärftliga sjukdom i de röda blodkropparna ger dock ett visst skydd mot malaria, som är eller var vanligt förekommande i de regioner där sjukdomen är vanlig. Denna selektiva överlevnadsfördel för bärare (känd som heterozygot fördel) är ansvarig för att mutationen hålls kvar i populationer långt över dess mutationsfrekvens. Bärare är heterozygota för thalassemiallelen, vilket innebär att endast en av deras två alleler är mutant (onormal). Det finns ett antal olika varianter av thalassemi. Var och en orsakas av en mutation i en annan position i arvsmassan.
I det avseendet liknar thalassemi en annan genetisk sjukdom som påverkar hemoglobin, sickle-cell-sjukdomen.
Det är möjligt att bota patienter med thalassemi med benmärgstransplantationer från kompatibla donatorer. Denna metod kräver dock en HLA-matchad kompatibel donator.
Bildgalleri
8 Bilder



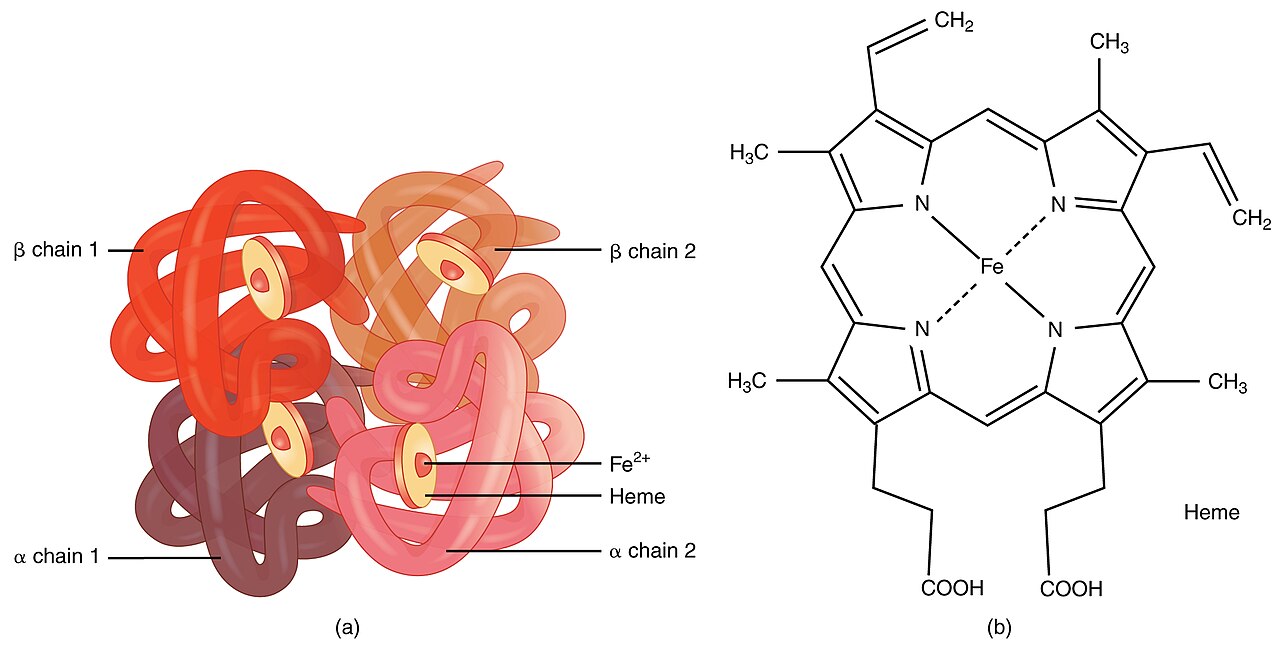
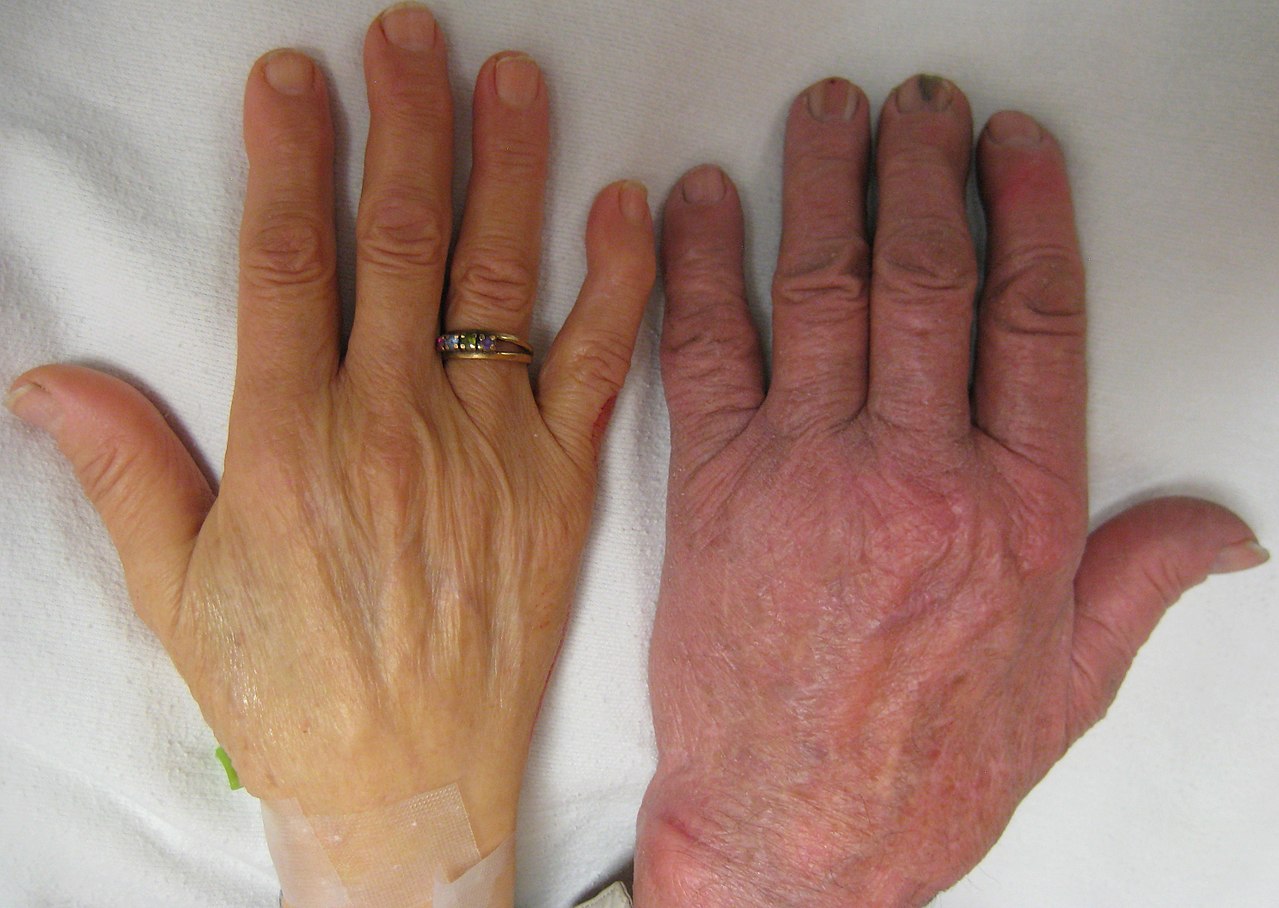


Typer av thalassemi
De två huvudtyperna är alfa-talassemi och beta-talassemi, beroende på vilka globinkedjor i hemoglobinet som påverkas. Var och en har flera undergrupper:
- Alfa-talassemi – orsakas av deletioner eller mutationer i gener som kodar alfa-globinkedjor. Kan vara allt från tyst bärare till svår sjukdom hos nyfödda (hydrops fetalis).
- Beta-talassemi – uppstår vid mutationer i beta-globingenen. Indelas ofta i beta-talassemi minor (bärare/trait), beta-talassemi intermedia och beta-talassemi major (Cooleys anemi), där major är den allvarligaste formen.
Symtom
Symtomen varierar med sjukdomens svårighetsgrad:
- Mild form (bärartillstånd): ofta inga eller lindriga symtom, upptäcks vid blodprov.
- Intermedia: trötthet, blekhet, återkommande infektionskänslighet, ljummen fysisk prestation.
- Svår form (major/Cooleys anemi): uttalad anemi, gulsot, tillväxthämning hos barn, benmissbildningar i ansikte och skalle, förstorad mjälte (splenomegali) och risk för hjärtsvikt om obehandlad.
Diagnos
Diagnos ställs med blodprover och genetiska tester:
- Fullständigt blodstatus (CBC) visar låg hemoglobin-nivå och förändrade RBC-index.
- Hemoglobinfraktionering eller hemoglobinanalys (t.ex. elektrofores) för att visa avvikande hemoglobintyper.
- Järnstatus (ferritin) för att särskilja från järnbristanemi.
- DNA-analyser för att påvisa specifika mutationer; viktigt för familjeutredning och prenatal diagnostik.
- Nyföddscreening och prenatala tester (korsprov, fostervattenprov, moderkaksprov) kan användas i högriskfamiljer.
Behandling
Behandlingen anpassas efter sjukdomens svårighetsgrad:
- Regelbundna blodtransfusioner för patienter med svår beta-talassemi för att upprätthålla normal hemoglobinnivå och förebygga hjärtskada.
- Järnkelering (järnbindande behandling) med läkemedel som desferrioxamin, deferasirox eller deferipron för att avlägsna överskott av järn som uppstår vid upprepade transfusioner.
- Folat (folsyra) som tillskott vid vissa fall för att stödja blodbildningen.
- Splenektomi (borttagning av mjälten) kan övervägas vid kraftig förstorad mjälte som orsakar ökad blodcellsförlust, men ökar infektionsrisk och kräver vaccinationer och ibland profylax.
- Benmärgstransplantation (allogen stamcellstransplantation) kan vara botande för utvalda patienter om en HLA-matcherad donator finns.
- Genterapi är ett snabbt växande forskningsfält; tekniker som lentivirala vektorer och CRISPR-baserad genredigering visar lovande resultat i kliniska prövningar och kan på sikt erbjuda kurativa alternativ för fler patienter.
- Övrig stödbehandling: vaccinationer, behandling av infektioner och behandling av komplikationer som hjärtsvikt eller hormonrubbningar.
Komplikationer
Vanliga och viktiga komplikationer att övervaka:
- Järnöverbelastning — lagras i lever, hjärta och endokrina organ och kan leda till levercirros, hjärtsvikt och diabetes om den inte behandlas.
- Benskelettförändringar — benmissbildningar och ökad benfrakturrisk p.g.a. benmärgshyperplasi.
- Infektioner — särskilt efter splenektomi; vaccination mot pneumokocker, meningokocker och Haemophilus influenzae rekommenderas.
- Endokrina störningar — tillväxthämning, försenad pubertet, hypotyreos, infertilitet p.g.a. järnläckage i körtlar.
Förebyggande och genetisk rådgivning
Genetisk rådgivning är viktig för par med ökad risk att få barn med svår thalassemi. Möjliga åtgärder inkluderar bärare‑screening innan graviditet, prenatala tester för att påvisa fostrets genotyp och rådgivning om reproduktionsalternativ (t.ex. preimplantatorisk genetisk diagnostik vid IVF).
Epidemiologi och prognos
Thalassemi förekommer framför allt i Medelhavsområdet, Mellanöstern, delar av Afrika, Indien och Sydostasien. På senare år har migration gjort att sjukdomen ses i många länder världen över. Prognosen har förbättrats avsevärt med modern transfusionsbehandling, järnkelering och specialistvård; många patienter lever numera långt in i vuxen ålder om de får adekvat uppföljning. Kort- och långtidssjukdomsresultat beror på tillgång till behandling, tidig diagnos och kontinuerlig vård.
Viktig information
Om du misstänker att du eller ett barn kan vara bärare eller ha thalassemi, kontakta primärvård eller specialist i hematologi för utredning. Vid bekräftad sjukdom är uppföljning vid ett center med erfarenhet av talassemi viktigt för att optimera behandling och förebygga komplikationer.
Frågor och svar
F: Vad är thalassemi?
S: Thalassemi är en genetisk sjukdom i blodet som har sitt ursprung i Medelhavsområdet. Sjukdomen orsakas av att de röda blodkropparna försvagas och förstörs på grund av muterade gener som påverkar hur kroppen tillverkar hemoglobin.
F: Vilka komplikationer är förknippade med thalassemi?
S: Komplikationer i samband med thalassemi kan vara lunginflammation, järnöverbelastning, benmissbildningar och kardiovaskulära sjukdomar.
F: Hur ger thalassemi ett skydd mot malaria?
S: Bärare av thalassemi har en selektiv överlevnadsfördel för bärare (så kallad heterozygot fördel) som bidrar till att hålla mutationen i populationer långt över dess mutationsfrekvens. Detta ger dem skydd mot malaria, som är eller var vanligt i regioner där detta drag är vanligt förekommande.
F: Finns det olika versioner av thalassemi?
S: Ja, det finns ett antal olika versioner av thalassemi som alla orsakas av en mutation i en annan position i arvsmassan. Den liknar en annan genetisk sjukdom som påverkar hemoglobin, sicklecellsjukdom.
F: Är det möjligt att bota patienter med thalassemi?
Svar: Ja, det är möjligt att bota patienter med thalassemi genom benmärgstransplantation från kompatibla donatorer som har en HLA-matchad kompatibel donator.
Relaterade artiklar
Författare
AlegsaOnline.com Thalassemi – ärftlig blodsjukdom: symtom, orsaker och behandling Leandro Alegsa
URL: https://sv.alegsaonline.com/art/97356
Källor
- accessmedicine.com : accessmedicine.com/content.aspx?aID=6123722
- mayoclinic.com : mayoclinic.com/health/thalassemia/DS00905/DSECTION=complications
- bloodjournal.hematologylibrary.org : HLA-matched sibling bone marrow transplantation for β-thalassemia major