Osteogenesis imperfecta (sprödbenssjukdom) – orsaker, typer & symtom
Osteogenesis imperfecta (sprödbenssjukdom) – läs om orsaker, typer, symtom, frakturrisk och hörselpåverkan. Fakta, diagnos och stöd för drabbade.
Osteogenesis imperfecta är en genetisk sjukdom som vanligen kallas för sprödbenssjukdom. Sjukdomen orsakas oftast av förändringar i de gener som bygger upp typ I-kollagen (vanligast i generna COL1A1 och COL1A2), vilket påverkar benens styrka och elasticitet. Det är ofta en autosomal dominant ärftlig sjukdom, vilket innebär att en person kan få sjukdomen om bara en av föräldrarna bär på den onormala genen. Det finns dock också former med autosomal recessiv ärftlighet och många fall uppstår genom nya (de novo) mutationer. Sjukdomen beskrevs först av Vrolik 1854.
Bildgalleri
10 Bilder







Orsaker och genetik
Osteogenesis imperfecta beror på defekter i produktionen eller strukturen av typ I-kollagen, ett viktigt protein i ben, senor och bindväv. De vanligaste orsakerna är mutationer i generna COL1A1 och COL1A2. Beroende på vilken mutation som förekommer påverkas mängden och kvaliteten på kollagenet, vilket ger allt från milda till mycket svåra symtom.
Typer (Sillence-klassificering I–IV)
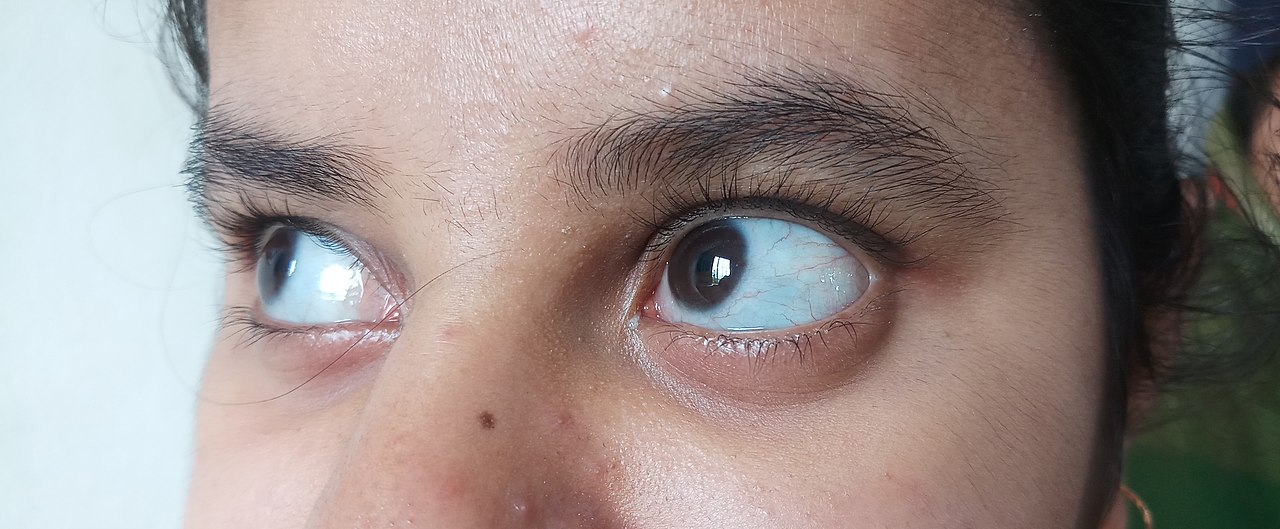
- Typ I – Den vanligaste och ofta lindrigaste formen. Personer har tendens till frakturer, särskilt i barndomen, samt tunnare/blåaktiga ögonvitor (blå sclera). Tandproblem (dentinogenesis imperfecta) kan förekomma. Normal eller nära normal längd.
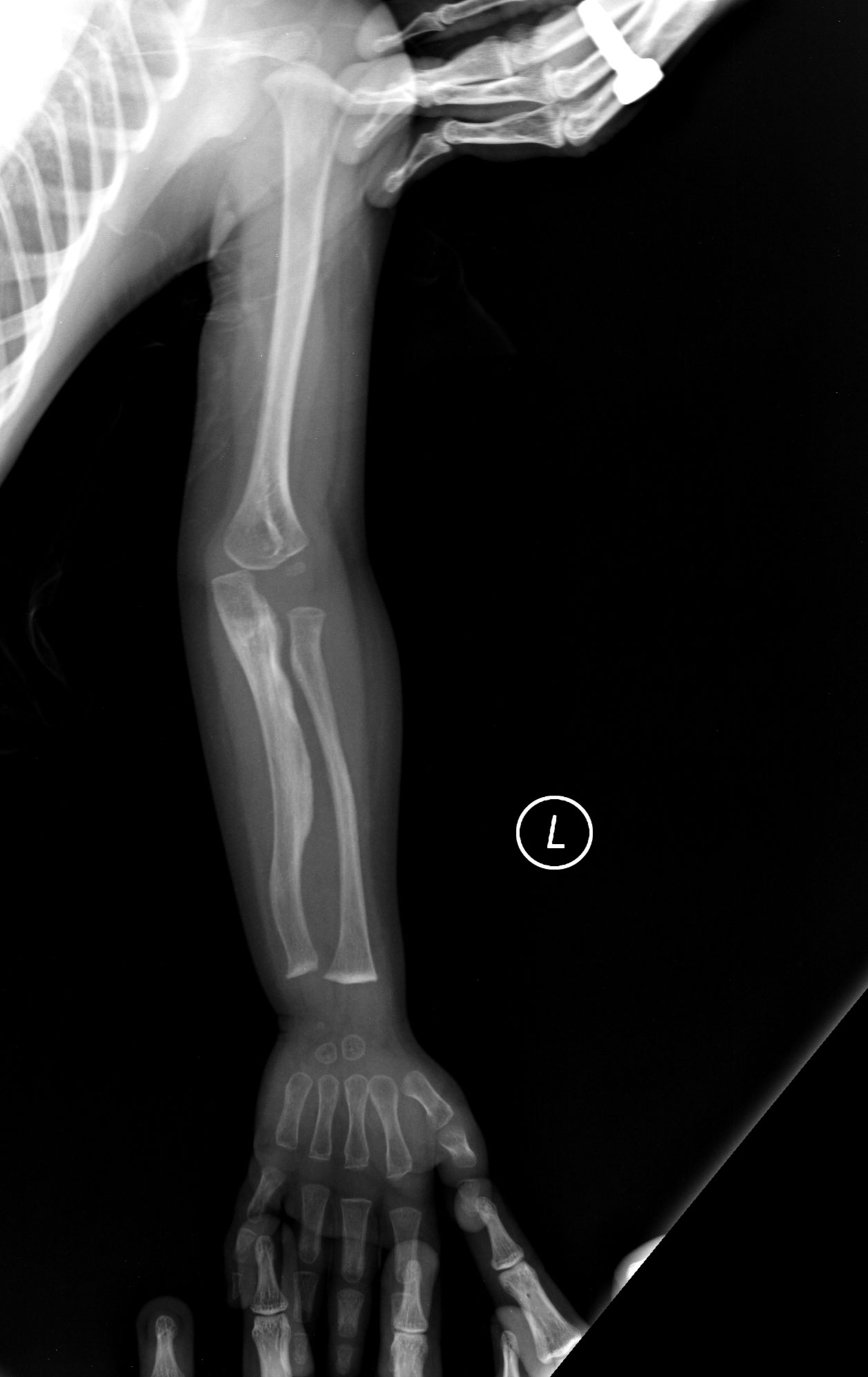
- Typ II – Den svåraste formen och ofta perinatalt dödlig. Kraftiga bentdeformiteter och upprepade frakturer redan i fosterstadiet eller vid födseln.
- Typ III – Progressiv, allvarlig form. Många frakturer före puberteten, uttalad kortvuxenhet, bendeformiteter och ofta nedsatt rörlighet. Ögonens färg kan vara blå, lila eller grå. Hörselnedsättning är vanlig.
- Typ IV – Måttligt svår form, med symtom som ligger mellan typ I och III. Sclera är oftast normalfärgade; tandproblem kan förekomma.
Det finns fler underkategorier och nyare genetiska klassifikationer, eftersom sjukdomen kan variera mycket även inom samma typ.
Vanliga symtom
- Upprepade benfrakturer, ofta redan i barndomen
- Blåaktiga eller missfärgade sclera (ögonvitor)
- Dentinogenesis imperfecta – sköra, missfärgade tänder
- Kortvuxenhet och ben-deformiteter (t.ex. skolios, kinkade långa ben)
- Ledinstabilitet och smärta
- Hörselnedsättning (förekommer hos cirka 50 % av vuxna med OI, kan börja i unga vuxna år)
- Andningsproblem vid svårare former (på grund av bröstkorgsdeformiteter)
Diagnos
Diagnosen ställs utifrån klinisk bild, röntgenfynd och ofta genetisk analys. Vanliga undersökningar:
- Röntgen av skelettet (visar typiska frakturmönster och deformiteter)
- Benmineraltäthet (DXA) för att bedöma benskörhet
- Genetisk analys för att påvisa mutation i relevanta gener (t.ex. COL1A1/COL1A2)
- Ultraljud eller fosterdiagnostik vid misstanke under graviditet om familjemutation är känd
- Öron- och tandläkarundersökning för att kartlägga följdtillstånd
Behandling och vård
Osteogenesis imperfecta kan inte botas, men många åtgärder kan minska problem och förbättra livskvaliteten. Behandlingen är ofta multidisciplinär:
- Medicinsk behandling: Bisfosfonater används ofta för att öka benmassa och minska frakturrisken, särskilt hos barn med svårare former. Kalcium- och D-vitamintillskott vid behov.
- Ortopedisk vård: Kirurgi med intramedullära stag (rodding) för att stabilisera långa ben och förebygga/åtgärda deformiteter. Operationer vid frakturer vid behov.
- Fysioterapi och rehabilitering: Träning för att stärka muskler, förbättra rörlighet och minska fallrisk. Hjälpmedel som ortoser, rullstol eller gånghjälpmedel kan behövas.
- Tandvård: Regelbundna kontroller och särskild tandbehandling vid dentinogenesis imperfecta.
- Hörselvård: Regelbundna hörselkontroller och tekniska hjälpmedel (hörapparat eller i vissa fall implantat).
- Psykosocialt stöd: Stöd från kurator, skolstöd och patientföreningar för att hantera vardagliga utmaningar.
Förebyggande och vardagsråd
- Lär dig och lär vårdare hur man förflyttar och hanterar ett barn med OI för att undvika onödiga frakturer (t.ex. rätt lyftteknik).
- Fallförebyggande åtgärder i hemmet — halkfria golv, säker förvaring, skyddande hjälmar vid hög risk för huvudskador i vissa fall).
- Anpassad fysisk aktivitet som stärker muskler utan att öka frakturrisken (t.ex. simning).
- Regelbunden tand- och hörselkontroll.
Graviditet, ärftlighet och rådgivning
Eftersom OI ofta är ärftligt är genetisk rådgivning viktig för familjer där någon är drabbad. Om den sjukdomsorsakande mutationen är känd kan man erbjuda prenatal diagnostik (kvalitativt fosterprov som fostervattensprov eller moderkaksprov) eller preimplantatorisk genetisk diagnostik (PGD) vid provrörsbefruktning, beroende på önskemål och lokal tillgång.
Prognos
Prognosen varierar mycket beroende på typ och svårighetsgrad. Många med lindrigare former (t.ex. typ I) har normal livslängd och god livskvalitet med rätt behandling. Svårare former kan ge tidig dödlighet, ofta på grund av andningsproblem eller komplicerade frakturer hos de mest allvarliga varianterna.
När kontakta vården
- Vid upprepade eller svåra frakturer utan tydlig orsak
- Om nyfödda med misstänkta deformiteter eller flera frakturer
- Vid hörselnedsättning, tandproblem eller andningssvårigheter hos någon med känd OI
Osteogenesis imperfecta kräver ofta långsiktig uppföljning av ett team med specialister (pediatriker, genetiker, ortoped, fysioterapeut, tandläkare och audiolog). Med rätt vård kan många drabbade leva aktiva och meningsfulla liv.
Symtom
Mindre allvarliga symtom på OI kan vara:
- lätt brutna ben
- lösa fogar
- Låg muskeltonus.
- blå, lila eller grå färg på den normalt vita delen av ögonen
- triangulär form i ansiktet
- tendens att utveckla skolios
- sköra tänder
OI har många andra allvarliga och dödliga symtom, bland annat andningsproblem och benmissbildningar.
Demografi
OI förekommer lika ofta hos män som kvinnor och kan drabba alla etniska grupper. OI uppstår i livmodern och det finns inget botemedel. Det upptäcks vanligtvis när någon bryter många ben; de kan få DNA-test för att kontrollera om de har OI. Det förekommer hos 1 av 20 000 födda barn.
Frågor och svar
F: Vad är osteogenesis imperfecta?
S: Osteogenesis imperfecta är en genetisk sjukdom som vanligen kallas benskörhet. Den försvagar eller förstör kollagenstaven, som ger benstyrka och leder till ben som är mer benägna att brytas.
F: Hur ärvs osteogenesis imperfecta?
S: Osteogenesis imperfecta är vanligtvis en autosomalt dominant sjukdom, vilket innebär att en person kan få den om bara en av föräldrarna har den onormala genen.
F: Vem identifierade först osteogenesis imperfecta?
S: Vrolik identifierade först osteogenesis imperfecta 1849.
F: Finns det något botemedel mot osteogenesis imperfecta?
S: Tyvärr finns det inget botemedel mot osteogenesis imperfecta.
F: Vilka är de fyra typerna av osteogenesis imperfecta?
S: De fyra typerna av osteogenesis imperfecta är typ ett, typ två, typ tre och typ fyra.
F: Vilka är symtomen på osteogenesis imperfecta typ tre?
S: Personer med typ tre av osteogenesis imperfecta kan ha mer än 100 frakturer före puberteten. Deras ögon får ofta en lila, blå eller grå nyans och personer med detta fall har också ofta hörselnedsättning.
F: Hur vanligt är det med hörselnedsättning hos personer med osteogenesis imperfecta?
S: 50 % av alla som har osteogenesis imperfecta har hörselnedsättning när de blir vuxna.
Relaterade artiklar
Författare
AlegsaOnline.com Osteogenesis imperfecta (sprödbenssjukdom) – orsaker, typer & symtom Leandro Alegsa
URL: https://sv.alegsaonline.com/art/73422
Källor
- oif.org : "Osteogenesis Imperfecta Foundation:"
- nlm.nih.gov : "Autosomal dominant: MedlinePlus Medical Encyclopedia"
- oif.org : "Osteogenesis Imperfecta Foundation: Understanding Bone Structure"