Molekylorbital – teori, egenskaper och användning
Översikt av molekylorbitaler: vad de är, hur de bildas, skillnader mot andra modeller, historik, exempel och praktiska tillämpningar inom kemi och beräkningar.
Molekylorbitaler (MO) är centrala begrepp i kvantkemin för att beskriva var elektronerna befinner sig i en molekyl och hur atomer binder till varandra. En molekylorbital är en matematisk vågfunktion som ger sannolikheten att hitta en elektron i olika regioner runt en molekyl. Begreppet hjälper kemister att tolka bindningsstyrka, magnetiska egenskaper och reaktivitet utifrån energinivåer och vågfunktioners form. För en introduktion till grundläggande termer, se grundläggande kemi.
Bildgalleri
5 Bilder
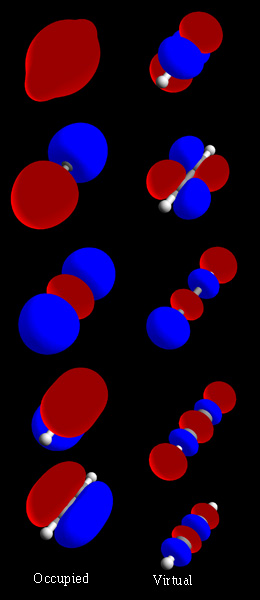



Hur molekylorbitaler bildas och tolkas
Molekylorbitaler konstrueras ofta genom att kombinera atomorbitaler i en metod kallad linjärkombination av atomorbitaler (LCAO). När två atomorbitaler överlappar kan de bilda en bindande orbital (med förstärkt elektronprobabilitet mellan kärnorna) eller en antibindande orbital (med en nod mellan kärnorna). Typiska symmetrier är sigma- och pi-orbitaler beroende på hur faserna av vågfunktionerna kombineras. Antalet molekylorbitaler som bildas motsvarar antalet ingående atomorbitaler, och elektronerna fyller dessa enligt Pauliprincipen och energiprincipen. Mer om elektronfördelning finns på elektronteori.
Matematisk och beräkningsmässig bakgrund
MOs är lösningar av Schrödingerekvationen för en molekyl i approximationer som Hartree–Fock eller tättfunktionalteori (DFT). Praktiskt arbete kräver ofta beräkningsmetoder och basuppsättningar för att approximera vågfunktioner; dessa ämnen beskrivs i detalj i facklitteraturen och i handledningar för teoretisk kemi och beräkningskemi. Tolkningen av orbitalernas form och energi ger information om bindningsordning, partiell laddningsfördelning och spektroskopiska övergångar.
Användningar och exempel
Molekylorbitalteorin förklarar till exempel varför H2 är stabilt (fylla en bindande σ-orbital), varför O2 är paramagnetiskt (obalans mellan π*-orbitaler) och hur konjugation i aromater leder till delokaliserade π-orbitaler. I organisk kemi används MO-begreppet för att förutsäga reaktionsförlopp i pericykliska reaktioner eller för att bedöma HOMO–LUMO-energiskillnader som styr fotokemiska egenskaper. För praktiska exempel och visualiseringar, se orbitalbilder och molekylberäkningar.
Historia och alternativa modeller
Molekylorbitalteorin utvecklades under 1900-talet av forskare som Robert Mulliken och Friedrich Hund som alternativa eller kompletterande sätt att förstå bindningar jämfört med valensbindningsteorin. Medan valensbindning fokuserar på parbildning mellan specifika atomorbitaler, beskriver MO-teorin elektroner som spridda över hela molekylen. Båda modellerna används idag beroende på vilken insikt eller noggrannhet som krävs; fler detaljer finns i historiska översikter på vetenskapshistoria och teoriöversikter.
Praktiska anmärkningar och resurser
- Molekylorbitaler har knutpunkter (noder) och faser som påverkar kemisk bindning; läs mer om nodstrukturer på kvantbegrepp.
- Beräkningsverktyg och program implementerar metoder som hartree–fock och DFT — startguider finns på beräkningspaket.
- Tillämpningar inom materialvetenskap, katalys och spektroskopi använder MO-analys för att förutsäga funktionella egenskaper — se exempel på tillämpningar och spektroskopi.
- För vidare läsning och utbildningsmaterial: introduktion, fördjupning och övningar.
Molekylorbitalteorin utgör en praktisk och teoretisk grund för modern kemi. Genom att kombinera experimentella observationer med beräkningsmetoder kan forskare och studenter tolka elektronfördelningar och förutsäga molekylers egenskaper med hög precision.

Historia
Ordet orbital användes för första gången på engelska av Robert S. Mulliken. Den tyske fysikern Erwin Schrödinger hade skrivit om MOs tidigare. Schrödinger kallade dem Eigenfunktion.
Fysikern Max Born beskrev teorin bakom molekylära banor 1926. I dag är den känd som Borns regel och ingår i Köpenhamnstolkningen av kvantmekaniken. När teorin ursprungligen föreslogs stämde den inte överens med Niels Bohrs atommodell. Bohrs modell beskrev elektroner som "kretsar" kring kärnan, då de rörde sig i cirklar. Men så småningom fick Bornmodellen populärt stöd eftersom den kunde beskriva elektronernas placering i molekyler och förklarade ett antal tidigare oförklarliga kemiska reaktioner.
Översikt
Atomorbitaler förutsäger elektronens position i en atom. Molekylära banor skapas när atomära banor sammanförs. En molekylorbital kan ge information om en molekyls elektronkonfiguration. Elektronkonfigurationen är den mest sannolika positionen och energin för en (eller ett par) elektron(er). Oftast representeras en MO som en linjär kombination av atomorbitaler (LCAO-MO-metoden), särskilt vid ungefärlig användning. Detta innebär att kemister antar att sannolikheten för att en elektron befinner sig på någon punkt i molekylen är summan av sannolikheterna för att elektronen befinner sig där baserat på de enskilda atomorbitalerna. LCAO-MO är en enkel modell för bindning i molekyler och är viktig för att studera molekylorbitalteori.
Teoretiska kemister använder datorer för att beräkna MOs för olika molekyler (både verkliga och imaginära). Datorn kan rita grafer av "molnet" för att visa hur sannolikt det är att elektronen kommer att befinna sig i ett visst område. Datorerna kan också ge information om molekylens fysiska egenskaper. De kan också säga hur mycket energi som krävs för att bilda molekylen. Detta hjälper kemister att säga om vissa små molekyler kan kombineras för att bilda större molekyler.
De flesta nuvarande metoder för beräkningskemi börjar med att beräkna MOs för ett system. Varje MO:s elektriska fält genereras av alla atomers kärnor och en genomsnittlig fördelning av de andra elektronerna.
Analogi
Att förstå MOs är som att veta var varje anställd befinner sig i en stor byggvaruhusbutik (utan att titta in i butiken). En analytiker vet hur många anställda som arbetar i butiken och vilken avdelning varje anställd tillhör. Han vet också att de anställda inte trampar varandra på tårna och att de anställda står i gången snarare än på varuhyllorna. Anställda lämnar sin egen avdelning för att hjälpa kunderna att hitta varor i andra avdelningar eller för att kontrollera lagerhållningen. En analytiker som anger var alla anställda i butiken befinner sig vid en viss tidpunkt utan att titta in i butiken är som en kemist som beräknar en molekyls MOs. På samma sätt som MOs inte kan ange den exakta platsen för varje elektron, är den exakta platsen för varje anställd inte känd. En MO som har ett nodalplan är som slutsatsen att de anställda går längs gångarna och inte genom hyllorna. Även om elektroner tillförs från en specifik atom, fyller elektronen en MO utan hänsyn till sin källatom. Detta är som att en anställd lämnar sin avdelning för att gå någon annanstans i butiken under dagen. En MO är alltså en ofullständig beskrivning av en elektron, på samma sätt som analytikerns beräkningar om den osynliga butiken är en ofullständig gissning om var de anställda befinner sig.

Bildande av molekylära orbitaler
Teoretiska kemister har uppfunnit regler för att beräkna MOs. Dessa regler bygger på en förståelse av kvantmekaniken. Kvantmekaniken hjälper kemisterna att använda vad fysiken säger om elektroner för att räkna ut hur elektronerna beter sig i molekyler. Molekylära banor bildas av "tillåtna" interaktioner mellan atomära banor. (Interaktionerna är "tillåtna" om symmetrierna (som bestäms av gruppteorin) hos atomorbitalerna är kompatibla med varandra). Kemister studerar atomorbitalernas växelverkan. Dessa interaktioner kommer från överlappningen (ett mått på hur väl två orbitaler interagerar konstruktivt med varandra) mellan två atomorbitaler. Överlappningen är viktig om atomorbitalerna ligger nära varandra i energi. Slutligen måste antalet MOs i en molekyl vara lika stort som antalet atomorbitaler i de atomer som förs samman för att bilda molekylen.
Kvalitativ metod
Kemister måste förstå geometrin hos MOs för att kunna diskutera molekylstruktur. LCMO-metoden (Linear combination of atomic orbitals molecular orbital) ger en grov men bra beskrivning av MOs. I denna metod uttrycks de molekylära orbitalen som linjära kombinationer av alla atomära orbitaler för varje atom i molekylen.
Linjära kombinationer av atomära orbitaler (LCAO)
Molekylära banor introducerades först av Friedrich Hund och Robert S. Mulliken 1927 och 1928.
Den linjära kombinationen av atomära banor eller "LCAO"-approximationen för molekylära banor introducerades 1929 av Sir John Lennard-Jones. Hans banbrytande artikel visade hur man kunde härleda fluor- och syremolekylernas elektroniska struktur från kvantprinciper. Detta kvalitativa tillvägagångssätt för teorin om molekylära orbitaler är en del av starten för den moderna kvantkemin.
Linjära kombinationer av atomära banor (LCAO) kan användas för att gissa vilka molekylära banor som bildas när molekylens atomer binds samman. I likhet med en atomorbital kan en Schrödingerekvation, som beskriver en elektrons beteende, också konstrueras för en molekylär orbital. Linjära kombinationer av atomära banor (summor och skillnader av atomära vågfunktioner) ger ungefärliga lösningar på de molekylära Schrödingerekvationerna. För enkla tvåatomiga molekyler representeras de vågfunktioner man får matematiskt av ekvationerna
Ψ = ca ψa + cb ψb
och
Ψ* = ca ψa - cb ψb
där Ψ och Ψ* är molekylära vågfunktioner för de bindande respektive antibindande molekylära orbitalerna, ψa och ψb är atomära vågfunktioner från atomerna a respektive b, och ca och cb är justerbara koefficienter. Dessa koefficienter kan vara positiva eller negativa, beroende på energierna och symmetrierna hos de enskilda atomorbitalerna. När de två atomerna närmar sig varandra överlappar deras atomära banor varandra och skapar områden med hög elektrontäthet. Det bildas alltså molekylära banor mellan de två atomerna. Atomerna hålls samman av den elektrostatiska attraktionen mellan de positivt laddade atomkärnorna och de negativt laddade elektronerna som upptar molekylära bindningsorbitaler.
Bindande, antibindande och icke-bindande MOs
När atomorbitaler interagerar kan den resulterande molekylära orbitalen vara av tre typer: bindande, antibindande eller icke-bindande.
Bindningsmetoder:
- Bindningsinteraktioner mellan atomorbitaler är konstruktiva (i fas) interaktioner.
- Bindande MOs har lägre energi än de atomorbitaler som kombineras för att skapa dem.
Antibonding MOs:
- Antibindningsinteraktioner mellan atomorbitaler är destruktiva (utanför fas) interaktioner.
- Antibonding MOs har högre energi än de atomära orbitaler som kombineras för att skapa dem.
Icke-bindande MOs:
- Icke-bindande MOs är resultatet av att det inte finns någon interaktion mellan atomära orbitaler på grund av brist på kompatibla symmetrier.
- Icke-bindande MOs har samma energi som atomorbitalerna för en av atomerna i molekylen.
HOMO och LUMO
Varje molekylorbital har sin egen energinivå. Kemister sorterar MO:erna efter energinivåer. Kemister antar att elektronerna kommer att fylla de MO:er som har den lägsta energinivån först. Om en molekyl till exempel har elektroner för att fylla 15 orbitaler, kommer de 15 MOs med de lägsta energinivåerna att fyllas. Den 15:e MO:n på listan skulle kallas "högst upptagna molekylära orbital" (HOMO) och den 16:e MO:n på listan skulle kallas "lägst oupptagna molekylära orbital" (LUMO). Skillnaden mellan HOMO:s energinivå och LUMO:s energinivå kallas bandgapet. Bandgapet kan ibland fungera som ett mått på molekylens excitabilitet: ju mindre energinivån är, desto lättare blir den exciterad. När elektronen exciteras hoppar den till en oockuperad MO. Detta kan till exempel hjälpa till att gissa om något kommer att avge ljus (luminescens).

Frågor och svar
Fråga: Vad är en molekylär orbit?
S: En molekylär orbital (eller MO) är en matematisk funktion som beskriver det vågliknande beteendet hos en elektron i en molekyl. Den förklarar vad som händer med elektroner när atomer förenas i en molekyl och kan ange sannolikheten för att hitta en elektron i ett visst område.
F: Hur bygger kemister upp matematiska modeller av molekylära banor?
S: Kemister bygger vanligtvis matematiska modeller av molekylära banor genom att kombinera atomära banor. Hybridbanor från varje atom i molekylen eller andra molekylära banor från grupper av atomer kan också användas. Datorer kan arbeta med dessa funktioner.
F: Vad har kvantmekaniken med studier av molekyler att göra?
S: Molekylära orbitaler gör det möjligt för kemister att tillämpa kvantmekanik för att studera molekyler. De besvarar frågor om hur atomerna i molekylerna håller ihop och ger en inblick i kemiska och fysikaliska egenskaper.
F: Vad är orbitaldiagram?
S: Orbitaldiagram är visuella representationer som visar var elektroner sannolikt skulle finnas i en atom utifrån dess olika rundade former.
F: Hur fungerar hybridorbitaler?
S: Hybridorbitaler kombinerar olika typer av atombanor till en ny typ som har unika egenskaper jämfört med sina beståndsdelar. Dessa hybrider används ofta när man bygger matematiska modeller för molekylära banor.
F: Hur kan datorer hjälpa till med att studera MOs?
S: Datorer kan bidra till att studera MOs genom att arbeta med deras funktioner och ge mer exakta förutsägelser eller förklaringar för kemiska och fysiska egenskaper i molekyler.
Relaterade artiklar
Författare
AlegsaOnline.com Molekylorbital – teori, egenskaper och användning Leandro Alegsa
URL: https://sv.alegsaonline.com/art/65861
Källor
- nobelprize.org : Nobelprize.org