Cystisk fibros (CF) – vad är det? Symtom, ärftlighet & behandling
Allt om cystisk fibros — symptom, ärftlighet, diagnos och moderna behandlingar. Lär dig hur sjukdomen påverkar vardagen och vilka medicinska alternativ som finns.
Cystisk fibros, även kallat mucoviscidos, ofta förkortat CF och ibland kallat "65 rosor" i vardagligt tal, är en ärftlig sjukdom. Den gör att kroppen bildar tjockt, klibbigt slem som lätt samlas i lungorna, i matsmältningssystemet och i andra organ. Detta slem gör det svårare att andas och kan orsaka upprepade infektioner och skador på lungvävnaden. Det finns inget botemedel mot cystisk fibros i dag, men det finns många mediciner och behandlingar som kan förbättra livet och förlänga överlevnaden.
Bildgalleri
4 Bilder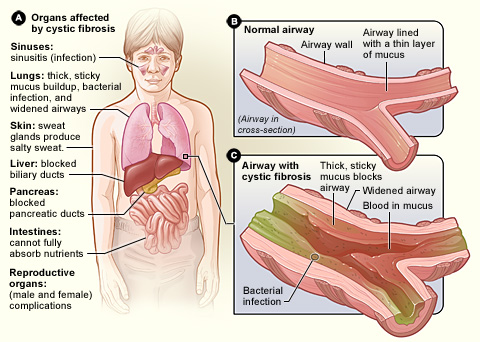

Vad orsakar cystisk fibros?
Cystisk fibros beror på förändringar i en gen som kallas CFTR (cystic fibrosis transmembrane conductance regulator). Sjukdomen är ärftlig och nedärvs enligt ett autosomalt recessivt mönster. Det betyder att ett barn måste få en sjuk gen från båda föräldrarna för att utveckla sjukdomen. En förälder kan bära på en muterad gen utan att själv ha symtom — då kallas personen bärare.
Ärftlighet och risker
- Om båda föräldrarna är bärare finns 25 % risk att ett barn får cystisk fibros, 50 % risk att barnet blir bärare utan sjukdom och 25 % chans att barnet varken blir bärare eller sjukt.
- Om endast en förälder är bärare kan barnet bli bärare men får inte sjukdomen.
- Genetisk rådgivning och testning kan ge besked om bärare-status, och det finns möjligheter till fosterdiagnostik och assisterad befruktning för par som vill ha det.
Symtom
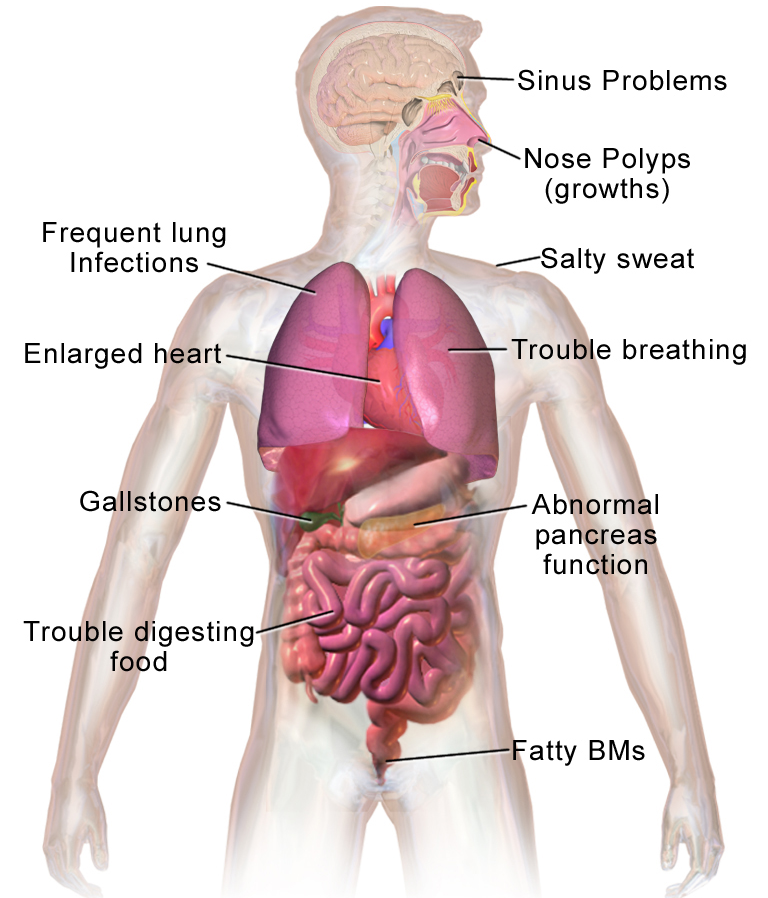
Symtomen varierar mycket i svårighetsgrad. Vissa får allvarliga problem redan i tidig barndom, andra får mildare symtom och upptäcks senare.
- Andningsvägar: långvarig hosta, slemhosta, upprepade luftvägsinfektioner, pipande andning och andfåddhet. Kronisk inflammation kan leda till bronkiektasier (utvidgade luftvägar).
- Matsmältning och näring: buksmärta, fettrik gråaktig avföring (steatorré), dålig viktuppgång eller tillväxt, brist på bukspottkörtel‐enzymer som leder till näringsbrist.
- Saltförlust och svettning: hög salthalt i svett som kan ge uttorkning och saltbrist, särskilt vid varm väderlek eller feber.
- Fertilitet: många män med CF är infertila på grund av frånvaro av bitestikeln eller vas deferens; kvinnor kan också ha nedsatt fertilitet men kan ofta bli gravida med rätt stöd.
- Övrigt: leverpåverkan, diabetes (CFRD), och hos vissa ben- och ledproblem.
Diagnos
- Nyföddscreening: många länder screenar nyfödda med ett blodprov som mäter immunreaktivt trypsinogen (IRT) och vid behov görs vidare genetiska tester.
- Svettest: mätning av klorid i svett är en viktig diagnosmetod — höga värden talar för CF.
- Genetisk testning: kartläggning av CFTR‐mutationer bekräftar diagnosen och kan vägleda behandling (vissa läkemedel fungerar bara vid vissa mutationer).
Behandling och vård
Behandlingen är mångsidig och syftar till att hålla luftvägarna öppna, förebygga infektioner, ge näring och behandla komplikationer. Vård ges oftast av ett multidisciplinärt team på en specialistklinik.
- Andningsbehandling: daglig fysioterapi för att rensa slem ur luftvägarna, andningsövningar och ibland andningshjälpmedel.
- Inhalationsbehandling: bronkdilaterare, slemlösande läkemedel som dornas alfa (DNase), inhalation av hyperton saltlösning för att lösa upp slem.
- Antibiotika: vid infektioner ges antibiotika peroralt, inhalerat eller intravenöst. Långtidsbehandling med inhalationsantibiotika används ofta för kronisk Pseudomonasinfektion.
- CFTR-modulatorer: nya läkemedel som potentiatorer och correctorer (t.ex. ivacaftor, lumacaftor/ivacaftor, tezacaftor/ivacaftor, elexacaftor/tezacaftor/ivacaftor) kan förbättra funktion av den felaktiga CFTR-proteinet hos personer med vissa mutationer.
- Mag-tarmstöd: pankreasenzymersättning för att hjälpa matsmältningen, vitamin A, D, E och K-tillskott samt energirik kost och ibland näringssond eller PEG vid behov.
- Kirurgiska åtgärder och transplantation: lungtransplantation kan bli aktuell vid mycket svårt lungsjukdom. I vissa fall behandlas lever- eller tarmkomplikationer kirurgiskt.
- Förebyggande åtgärder: vaccinationer, tidig behandling av infektioner och god infektionskontroll. Personer med CF uppmanas ofta att undvika nära kontakt med andra personer med CF för att minska risken för korsspridning av resistenta bakterier.
Smittar cystisk fibros?
Nej. Cystisk fibros är en genetisk sjukdom och kan inte smitta från person till person. Däremot kan bakterier och andra infektioner som drabbar personer med CF överföras mellan människor, vilket är anledningen till att särskilda hygien- och avståndsrutiner ofta rekommenderas inom CF-vården.
Prognos och livskvalitet
Prognosen för personer med cystisk fibros har förbättrats kraftigt under de senaste decennierna tack vare bättre behandlingar, screeningar och nya läkemedel. Många lever långt in i vuxen ålder och kan arbeta, studera och bilda familj. Tidig upptäckt, regelbunden specialistvård och anpassad behandling förbättrar ofta både livslängd och livskvalitet.
Vid behov av mer information
Om du misstänker att du eller ditt barn kan ha cystisk fibros, eller om du vill veta mer om genetisk rådgivning, kontakta din vårdcentral eller en specialistmottagning för cystisk fibros. De kan erbjuda tester, rådgivning och remiss till rätt vårdteam.
Vad CF gör med kroppen
Cystisk fibros påverkar hela kroppen. Överlag har kroppen problem med att flytta salt till de delar av kroppen som behöver det. Eftersom kroppen har problem med att flytta salt samlas det på ställen där det inte ska finnas, som i lungorna, magen och tarmarna.
Lungor
När salt fastnar i lungorna leder det till att det blir mindre vatten i lungorna, vilket gör att slemmet blir mycket tjockt. Det blir mycket svårt att andas. Behandlingen av detta är andningsmedicin som hjälper till att tillföra vatten till lungorna för att hålla slemmet tunnare så att det blir lättare att hosta upp. När det finns tunnare och mindre slem är det lättare att andas.
Behandling
Det finns inget botemedel mot cystisk fibros. Även om man kan göra saker för att hålla sig frisk. Hälsosamma vanor hindrar personen från att bli mer sjuk. Människor kan hålla sig rena. Människor kan hålla sig borta från bakterier. De kan dricka vatten för att hjälpa slemmet att försvinna. Att ta enzymer hjälper till att smälta maten om det finns slem i magen.
Motion rensar slem. Den bygger starka muskler och ben och stärker lungorna. Att ta vitaminer hjälper kroppen att bekämpa infektionen. Det hjälper också kroppen att växa och fungera väl.
- Inhalationsantibiotika används för att förhindra att bakterier växer i det tjocka slemmet.
- Inhalerat saltvatten hjälper till att hålla lungorna fuktiga.
Testning för cystisk fibros
- Svettkloridtest - testar saltnivån i en persons svett.
- Genetiskt test - detta används om svettprovet är positivt för att se om de har båda generna.
65 rosor
"65 rosor" är vad vissa barn kallar sitt tillstånd eftersom cystisk fibros är svårt för ett litet barn att säga. "65 rosor" är också en fras som Cystic Fibrosis Foundation har registrerat som varumärke för att kontrollera användningen. Det är ett mycket hjälpsamt sätt för små barn att förstå. När den uttalas högt låter den ungefär som cystisk fibros.
Frågor och svar
F: Vad är cystisk fibros?
S: Cystisk fibros är ett tillstånd som gör att kroppen producerar tjockt, klibbigt slem som kan byggas upp i lungorna, matsmältningssystemet och andra delar av kroppen.
F: Hur orsakas cystisk fibros?
S: Cystisk fibros orsakas av att man ärver cystisk fibros-genen från båda föräldrarna.
F: Kan en person med bara en cystisk fibros-gen föra sjukdomen vidare till sitt barn?
S: En person med bara en cystisk fibros-gen kanske inte själv har sjukdomen, men kan ändå överföra genen till sitt barn.
F: Är cystisk fibros smittsam?
S: Nej, cystisk fibros är inte smittsamt och kan inte överföras från en person till en annan.
F: Finns det något botemedel mot cystisk fibros?
S: Nej, det finns för närvarande inget botemedel mot cystisk fibros, men det finns många mediciner som kan hjälpa till att hantera tillståndet.
F: Vilka delar av kroppen kan påverkas av cystisk fibros?
S: Cystisk fibros kan påverka lungorna, matsmältningssystemet och andra delar av kroppen.
F: Hur kan personer med cystisk fibros hålla sig friska?
S: Personer med cystisk fibros kan hålla sig friska genom att ta medicin, övervaka sitt tillstånd och följa en hälsosam livsstil.
Relaterade artiklar
Författare
AlegsaOnline.com Cystisk fibros (CF) – vad är det? Symtom, ärftlighet & behandling Leandro Alegsa
URL: https://sv.alegsaonline.com/art/24952