Pulmonell arteriell hypertension (PAH)
Sällsynt sjukdom med förhöjt blodtryck i lungornas artärer som ger andfåddhet, trötthet och risk för hjärtsvikt; diagnos kräver specialistutredning och riktad behandling.
Pulmonell arteriell hypertension (förkortat PAH) är en form av pulmonell hypertension där trycket i lungornas artärer är förhöjt. Tillståndet påverkar blodflödet i lungorna och ökar belastningen på höger kammare i hjärtat. Det kan utvecklas gradvis och leda till svår andnöd och nedsatt fysisk kapacitet. Begreppet används ofta synonymt med svår sjukdom i lungartärernas små kärl och skiljer sig från andra orsaker till högt lungtryck.
Bildgalleri
9 Bilder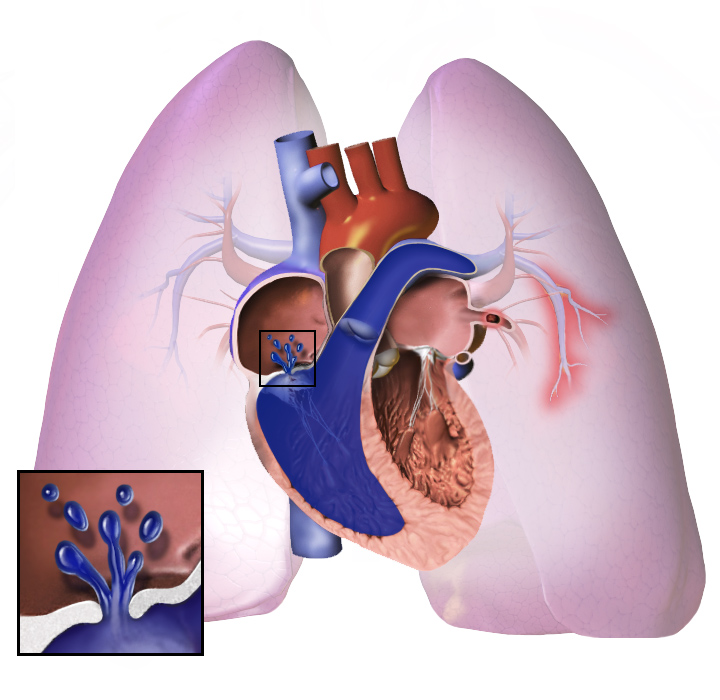






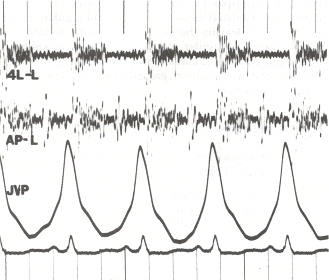
Orsaker och patofysiologi
PAH beror på en kombination av vasokonstriktion, inflammation, trombos i situ och strukturell ombyggnad (remodellering) av små lungartärer. Dessa förändringar minskar kärlens lumen och ökar motståndet mot blodflödet. Orsakerna kan vara idiopatiska, ärftliga (till exempel varianter i BMPR2-genen), kopplade till läkemedel eller toxiner, eller associerade med andra sjukdomar som bindvävssjukdomar, HIV-infektion, portahypertension eller medfödda hjärtfel. Vissa former är direkt utlösbara av specifika läkemedel eller droger.
Symtom
- Förvärrad andfåddhet vid ansträngning och successivt även i vila; svårigheter att andas.
- Trötthet och minskad uthållighet.
- Yrsel och i vissa fall synkope vid ansträngning.
- Bröstsmärta, hjärtklappning och tecken på vätskeansamling i benen.
- Vissa patienter behöver extra syre i avancerade skeden.
Utredning
Utredning inleds ofta med arbets- eller vila-ekokardiografi som visar tecken på förhöjt tryck i lungkretsloppet. Definitiv diagnos kräver invasivt mätning med högerhjärtkateterisering för att fastställa trycknivåer och motstånd. Ytterligare undersökningar kan innefatta lungfunktionsprov, ventilations-/perfusion-scintigrafi, datortomografi, blodprover och genetisk analys vid misstanke om ärftlighet.
Behandling och prognos
Behandlingen kombinerar allmänna åtgärder och läkemedel riktade mot de mekanismer som driver sjukdomen. Basåtgärder kan vara fysisk anpassning, vaccinationer, diuretika vid vätskeöverskott och ibland antikoagulantia. Modern läkemedelsbehandling innefattar tre huvudklasser: prostacyklinanaloger, endotelinreceptorantagonister och läkemedel som påverkar NO–cGMP-vägen (till exempel PDE5-hämmare och stimulatorer av lösligt guanylatcyklas). I avancerade fall kan parenteral prostacyclinbehandling eller kombinationsbehandling vara nödvändig.
Trots bättre behandlingar är sjukdomen allvarlig och kan leda till progressiv hjärtsvikt; för vissa patienter övervägs lungtransplantation eller hjärt-lungtransplantation. Tidig diagnos och behandling förbättrar livskvalitet och överlevnad.
Notable fakta och epidemiologi
PAH är en sällsynt sjukdom med störst förekomst hos yngre till medelålders kvinnor. Kunskapen om orsaker och behandling har utvecklats kraftigt under senare decennier, men tillståndet kräver ofta vård vid specialiserade centra för pulmonell hypertension. Patienter bör följa riktlinjer och uppföljning hos multidisciplinära team för bästa resultat.
För mer generella fakta om blodtryck i lungkretsloppet, se även information om blodtryck och lungkretsloppets anatomi.
Tecken och symtom
Personer med pulmonell hypertension har svårt att andas. De blir också lätt trötta. En del av dem svimmar också lätt. De kan ha smärta i bröstet. Vissa patienter har svullnad i fötter och vrister. Dessa symtom förvärras vid motion eller hårt arbete.
Eftersom många sjukdomar kan göra det svårt att andas måste läkaren ta reda på patientens bakgrund. Detta hjälper läkaren att behandla patienten, även om patienten har en annan sjukdom. Läkaren gör också flera tester. Pulmonell hypertension gör att hjärtat låter annorlunda. Ett test är att mäta blodtrycket inne i lungartären, det blodkärl som går från hjärtat till lungorna.
För att fastställa orsaken gör läkaren i allmänhet en grundlig anamnes. En detaljerad familjehistoria tas fram för att avgöra om sjukdomen kan vara familjär. En historia av exponering för kokain, metamfetamin, alkohol som leder till skrumplever och rökning som leder till emfysem anses vara betydelsefull. Fysisk undersökning utförs för att leta efter typiska tecken på pulmonell hypertension, inklusive ett högt P2-ljud (ljudet av pulmonell klaffens stängning), (para)sternal heave, spridning av jugularvenen, fotödem, ascites, hepatojugulärt återflöde, klubbning etc.
Vad som går fel med kroppen
Vid pulmonell hypertension blir blodkärlen i lungorna för trånga. Blodtrycket i lungorna blir högt. Hjärtat arbetar mycket hårt för att pumpa blod genom de trånga blodkärlen. Senare blir blodkärlen i lungorna hårda och tjocka. Hjärtat måste arbeta hårdare.
Hjärtat kan arbeta så hårt att det blir sjukt. Detta kallas hjärtsvikt. Det sjuka hjärtat kan inte pumpa blodet på ett bra sätt. Mindre blod går till lungorna, så blodet får mindre syre. Detta gör det svårt att andas. Detta blir värre när man tränar eller arbetar hårt.
Orsaker
Den vanligaste orsaken till pulmonell hypertension är vänster hjärtsvikt. Detta orsakar pulmonell venös hypertension. Detta leder till lungödem eller vätskeansamling i lungorna.
Många sjukdomar kan orsaka pulmonell arteriell hypertension (PAH).
- Lungsjukdomar som gör att blodet får mindre syre, t.ex:
· kronisk obstruktiv lungsjukdom (COPD)
· Interstitiell lungsjukdom.
· Pickwickiskt syndrom
- problem med immunförsvaret, t.ex:
· AIDS
· sklerodermi
· Andra autoimmuna sjukdomar.
- Leverproblem.
· skrumplever
· Portal hypertension.
- andra orsaker
· sömnapné
· ta tabletter för att gå ner i vikt, t.ex. Fen-Phen, Aminorex, fenfluramin (Pondimin) och fentermin.
· Medfödd hjärtsjukdom.
· Sköldkörtelsjukdomar,
· tar droger som kokain
· möjligen Human herpesvirus 8
När en person har pulmonell hypertension utan någon annan orsak kallas detta idiopatisk pulmonell arteriell hypertension (IPAH).
När det finns en familjehistoria kallas sjukdomen för familjär pulmonell arteriell hypertension (FPAH). IPAH och FPAH anses nu vara genetiska sjukdomar som är kopplade till mutationer i BMPR2-genen, som kodar för en receptor för benmorfogenetiska proteiner, samt 5-HT(2B)-genen, som kodar för en serotoninreceptor.
Inom medicinen är pulmonell hypertoni (PH) en ökning av blodtrycket i lungartären eller lungkärlen, vilket leder till andnöd, yrsel, svimning och andra symtom, som alla förvärras av ansträngning. Beroende på orsaken kan pulmonell hypertoni vara en allvarlig sjukdom med en markant minskad träningstolerans och högersidig hjärtsvikt. Den identifierades först av dr Ernst von Romberg 1891. Det kan vara en av fem olika typer, arteriell, venös, hypoxisk, tromboembolisk eller diverse.
Även om termerna primär pulmonell hypertoni (dvs. av okänd orsak) och sekundär pulmonell hypertoni (dvs. på grund av ett annat medicinskt tillstånd) fortfarande förekommer i material som sprids till patienter och allmänheten, har dessa termer i stort sett övergivits i den medicinska litteraturen. Denna förändring har skett eftersom den äldre dikotoma klassificeringen inte återspeglade patofysiologi eller resultat. Den ledde till felaktiga terapeutiska beslut, dvs. att endast behandla "primär" pulmonell hypertension. Detta ledde i sin tur till terapeutisk nihilism för många patienter som betecknades som "sekundär" pulmonell hypertoni och kan ha bidragit till deras död. Termen "primär pulmonell hypertension" har nu ersatts av "idiopatisk pulmonell arteriell hypertension". Termerna "primär" och "sekundär" pulmonell hypertension bör inte längre användas. Ytterligare information finns i avsnittet Klassificering nedan.
Orsaker
Den vanligaste orsaken till pulmonell hypertension är vänster hjärtsvikt som leder till pulmonell venös hypertension. Detta kan bero på systolisk eller diastolisk funktionsstörning i vänster kammare eller på klaffdysfunktion, t.ex. mitralregurgitation eller mitralstenos. Det visar sig vanligtvis som lungödem.
Vanliga orsaker till pulmonell arteriell hypertoni (PAH) är bland annat hiv, sklerodermi och andra autoimmuna sjukdomar, skrumplever och portal hypertoni, sicklecellsjukdom, medfödd hjärtsjukdom och sköldkörtelsjukdomar. Användning av viktminskningspiller som Fen-Phen, Aminorex, fenfluramin (Pondimin) och fentermin ledde tidigare till utveckling av PAH.
Patogenes
Oavsett den ursprungliga orsaken innebär pulmonell hypertension att blodkärlen i och i lungorna dras åt. Detta gör det svårare för hjärtat att pumpa blod genom lungorna, på samma sätt som det är svårare att få vatten att rinna genom ett smalt rör än ett brett. Med tiden blir de drabbade blodkärlen både styvare och tjockare, vilket ytterligare ökar blodtrycket i lungorna och försämrar blodflödet. Dessutom orsakar den ökade arbetsbelastningen på hjärtat förtjockning och utvidgning av höger kammare, vilket gör att hjärtat har sämre förmåga att pumpa blod genom lungorna, vilket orsakar höger hjärtsvikt. När blodflödet genom lungorna minskar får den vänstra sidan av hjärtat mindre blod. Detta blod kan också innehålla mindre syre än normalt. Därför blir det allt svårare för hjärtats vänstra sida att pumpa för att tillföra tillräckligt med syre till resten av kroppen, särskilt vid fysisk aktivitet.
Diagnos
Eftersom pulmonell hypertoni kan vara av fem olika typer måste en rad tester utföras för att skilja pulmonell arteriell hypertoni från venösa, hypoxiska, tromboemboliska eller andra typer av hypertoni.
En fysisk undersökning görs för att leta efter typiska tecken på pulmonell hypertension. Dessa omfattar förändrade hjärtljud, t.ex. ett brett uppdelat S2 eller andra hjärtljud, ett högt P2 eller lungklaffens stängningsljud (en del av det andra hjärtljudet), (para)sternal heave, eventuellt S3 eller tredje hjärtljud och pulmonell regurgitation. Andra tecken är bl.a. jugular venous distension (utvidgning av jugularvenerna), perifert ödem (svullnad av fotleder och fötter), ascites (buksvullnad på grund av ansamling av vätska), hepatojugulärt återflöde och klubbning.
Ytterligare åtgärder krävs för att bekräfta förekomsten av pulmonell hypertension och utesluta andra möjliga diagnoser. Dessa omfattar i allmänhet lungfunktionsundersökningar, blodprov, elektrokardiografi (EKG), mätning av arteriella blodgaser, röntgenbilder av bröstkorgen (följt av högupplösande datortomografi om interstitiell lungsjukdom misstänks) och ventilation-perfusion eller V/Q-skanning för att utesluta kronisk tromboembolisk pulmonell hypertension. Biopsi av lungan är vanligtvis inte indicerad om inte den pulmonella hypertensionen tros bero på en underliggande interstitiell lungsjukdom. Lungbiopsier är dock behäftade med blödningsrisker på grund av det höga intrapulmonala blodtrycket. Den kliniska förbättringen mäts ofta genom ett "sexminuters gångtest", dvs. den sträcka som patienten kan gå på sex minuter. Stabilitet och förbättring i detta mått korrelerar med bättre överlevnad.
Även om det pulmonella arteriella trycket kan uppskattas med hjälp av ekokardiografi ger tryckprovtagning med en Swan-Ganz-kateter den mest säkra mätningen. PAOP och PVR kan inte mätas direkt med ekokardiografi. För att diagnostisera PAH krävs därför en hjärtkateterisering. En Swan-Ganz-kateter kan också mäta hjärtminutvolymen, som är mycket viktigare för att mäta sjukdomens svårighetsgrad än det pulmonella arteriella trycket.
Det normala pulmonella arteriella trycket hos en person som lever på havsnivå har ett medelvärde på 12-16 mm Hg (1600-2100 Pa). Definitiv pulmonell hypertension föreligger när medeltrycket i vila överstiger 25 mm Hg (3300 Pa). Om medeltrycket i lungartären stiger över 30 mm Hg (4000 Pa) vid ansträngning betraktas detta också som pulmonell hypertension.
För att PAH ska kunna diagnostiseras krävs förekomst av pulmonell hypertension och två andra tillstånd. Pulmonalartärens ocklusionstryck (PAOP eller PCWP) måste vara mindre än 15 mm Hg (2000 Pa) och pulmonal kärlmotstånd (PVR) måste vara större än 3 Wood-enheter (240 dyn-s-cm−5 eller 2,4 mN-s-cm−5 ).
Klassificering
Nuvarande klassificering
År 2003 sammankallades det tredje världssymposiet om pulmonell arteriell hypertoni i Venedig för att ändra klassificeringen utifrån den nya förståelsen av sjukdomsmekanismerna. Det reviderade system som denna grupp utvecklade utgör den nuvarande ramen för att förstå pulmonell hypertension.
Systemet innehåller flera förbättringar jämfört med det tidigare klassificeringssystemet Evian Classification från 1998. Beskrivningarna av riskfaktorer har uppdaterats och klassificeringen av medfödda systemiska-till-pulmonala shuntar har reviderats. En ny klassificering av genetiska faktorer vid PH rekommenderades, men genomfördes inte eftersom tillgängliga uppgifter bedömdes vara otillräckliga.
Det reviderade klassificeringssystemet från Venedig 2003 kan sammanfattas på följande sätt:
- WHO-grupp I - Pulmonell arteriell hypertension (PAH)
- WHO-grupp II - Pulmonell hypertoni i samband med vänster hjärtsjukdom
- WHO-grupp III - Pulmonell hypertension i samband med lungsjukdomar och/eller hypoxemi.
- WHO-grupp IV - Pulmonell hypertension på grund av kronisk trombotisk och/eller embolisk sjukdom.
- WHO-grupp V - Diverse
Föregående terminologi
Tidigare användes begreppen primär och sekundär pulmonell hypertension (PPH och SPH) för att klassificera sjukdomen. Detta ledde till antagandet att endast den primära sjukdomen bör behandlas och att den sekundära varianten bör ignoreras till förmån för behandling av endast den underliggande sjukdomen. I själva verket kan alla former av pulmonell arteriell hypertoni behandlas. Tyvärr finns detta klassificeringssystem fortfarande kvar i många läkares medvetande och leder troligen till att många patienter nekas behandling. Denna nihilistiska inställning till pulmonell arteriell hypertoni kan också bidra till underdiagnostik. Man uppskattar att det finns omkring 100 000 patienter med PAH i USA, men endast 15-20 000 har fått en diagnos. Många andra har feldiagnostiserats som KOL, astma eller hjärtsvikt.
Termen primär pulmonell hypertension (PPH) har nu ersatts av idiopatisk pulmonell arteriell hypertension (IPAH) i en stor del av den medicinska litteraturen. Vissa läkare fortsätter dock att använda den äldre klassificeringen på ett olämpligt sätt.
Epidemiologi
IPAH är en sällsynt sjukdom med en incidens på cirka 2-3 per miljon per år och en prevalens på cirka 15 per miljon. Kvinnor har nästan tre gånger så stor sannolikhet att drabbas av IPAH som män.
Andra former av PAH är mycket vanligare. Vid sklerodermi har förekomsten uppskattats till 6-60 % av alla patienter, vid reumatoid artrit upp till 21 %, vid systemisk lupus erythematosus 4-14 %, vid portal hypertension mellan 2 och 5 %, vid hiv cirka 0,5 % och vid sicklecellsjukdom mellan 20 och 40 %.
Bantningspiller som Fen-Phen har en årlig incidens på 25-50 per miljon per år.
Behandling
Behandlingen bestäms av om PH är arteriell, venös, hypoxisk, tromboembolisk eller annan. Eftersom pulmonell venös hypertoni är synonymt med kongestiv hjärtsvikt är behandlingen att optimera vänsterkammarfunktionen genom användning av diuretika, betablockerare, ACE-hämmare etc. eller att reparera/byta mitralisklaffen eller aortaklaffen.
Vid PAH anses livsstilsförändringar, digoxin, diuretika, orala antikoagulantia och syrgasbehandling vara konventionell behandling, men har aldrig visat sig vara fördelaktigt i en randomiserad, prospektiv studie.
Högdos kalciumkanalblockerare är användbara för endast 5 % av IPAH-patienterna som är vasoreaktiva med Swan-Ganz-katetern. Tyvärr har kalciumkanalblockerare i stor utsträckning missbrukats och förskrivits till många patienter med icke vasoreaktiv PAH, vilket leder till överdriven morbiditet och mortalitet.
Vasoaktiva ämnen
Tre huvudvägar är inblandade i onormal proliferation och kontraktion av de glatta muskelcellerna i lungartären hos patienter med pulmonell arteriell hypertension. Dessa vägar motsvarar viktiga terapeutiska mål i detta tillstånd och spelar en roll när det gäller att avgöra vilken av tre klasser av läkemedel som ska användas - endotelinreceptorantagonister, fosfodiesteras typ 5-hämmare och prostacyklinderivat.
Prostacyklin (prostaglandin I2 ) anses allmänt vara den mest effektiva behandlingen av PAH. Epoprostenol (syntetiskt prostacyklin, marknadsförs som Flolan®) ges via kontinuerlig infusion som kräver en halvpermanent central venkateter. Detta administreringssystem kan orsaka sepsis och trombos. Flolan® är instabil och måste därför förvaras på is under administreringen. Eftersom det har en halveringstid på 3-5 minuter måste infusionen vara kontinuerlig (dygnet runt), och ett avbrott kan vara dödligt. Andra prostanoider har därför utvecklats. Treprostinil (Remodulin®) kan ges intravenöst eller subkutant, men den subkutana formen kan vara mycket smärtsam. Iloprost (Ilomedin®) används också i Europa intravenöst och har en längre halveringstid. Iloprost (marknadsförs som Ventavis®) är den enda inhalerade formen av prostacyklin som är godkänd för användning i USA och Europa. Denna administreringsform har fördelen att den selektivt deponeras i lungorna med färre systemiska biverkningar.
Den dubbla (ETA och ETB ) endotelinreceptorantagonisten bosentan (som marknadsförs som Tracleer®) godkändes 2001. Två selektiva endotelinreceptorantagonister (endast ETA ) befinner sig i slutskedet av godkännandet: sitaxsentan och ambrisentan. Sildenafil, en selektiv hämmare av cGMP-specifikt fosfodiesteras typ 5 (PDE5), godkändes för behandling av PAH 2005. Det marknadsförs för PAH som Revatio®. Tadalafil (som för närvarande marknadsförs som Cialis® för erektil dysfunktion) befinner sig för närvarande i fas III-studier. Vasoaktiv intestinal peptid genom inhalation bör börja kliniska prövningar för PAH 2007. PRX-08066 är en serotoninantagonist som för närvarande utvecklas för hypoxisk pulmonell hypertension.
Kirurgiskt
Förmaksseptostomi är ett kirurgiskt ingrepp som skapar en kommunikation mellan höger och vänster förmak. Det lindrar trycket på höger sida av hjärtat, men till priset av lägre syrehalter i blodet (hypoxi). Det utförs bäst på erfarna centra. Lungtransplantation botar pulmonell arteriell hypertension, men lämnar patienten med de komplikationer som transplantationen medför och en överlevnad på cirka fem år.
Pulmonell tromboendarterektomi (PTE) är ett kirurgiskt ingrepp som används vid kronisk tromboembolisk pulmonell hypertension. Det är ett kirurgiskt avlägsnande av en organiserad tromb (koagel) tillsammans med slemhinnan i lungartären; det är ett stort och mycket svårt ingrepp som för närvarande utförs på några få utvalda centra. Fallserier visar på anmärkningsvärda framgångar hos de flesta patienter.
Behandling av hypoxisk och diverse varianter av pulmonell hypertension har inte fastställts. För närvarande pågår dock studier av flera medel som omfattar patienter. Många läkare kommer att behandla dessa sjukdomar med samma läkemedel som för PAH, tills bättre alternativ blir tillgängliga.
Prognos
NIH:s IPAH-register från 1980-talet visade en obehandlad medianöverlevnad på 2-3 år från diagnosen, och dödsorsaken var vanligen högerkammarsvikt (cor pulmonale). Även om denna siffra ofta citeras är den förmodligen irrelevant i dag. Resultaten har förändrats dramatiskt under de senaste två decennierna. Detta kan bero på nyare läkemedelsbehandling, bättre allmän vård och tidigare diagnos (lead time bias). En nyligen genomförd resultatstudie av de patienter som hade påbörjat behandling med bosentan (Tracleer®) visade att 86 % av patienterna levde efter tre år. Eftersom flera medel nu finns tillgängliga används kombinationsbehandling i allt större utsträckning. Man vet inte hur dessa medel påverkar överlevnaden, eftersom många av dem har utvecklats först nyligen. Det skulle inte vara orimligt att förvänta sig att medianöverlevnaden kommer att överstiga 10 år inom en snar framtid.
Frågor och svar
F: Vad är pulmonell hypertension eller PH?
S: Pulmonell hypertension eller PH är ett tillstånd där det finns ett högt blodtryck i lungorna.
F: Vilka är symtomen på pulmonell hypertension?
S: Symtomen på pulmonell hypertension inkluderar andningssvårigheter, yrsel, trötthet och svimning.
F: Varför behöver vissa personer med pulmonell hypertension extra syre?
S: Vissa personer med pulmonell hypertension behöver extra syre eftersom tillståndet gör det svårt för dem att andas.
F: När blir symtomen på pulmonell hypertension värre?
S: Symtomen på pulmonell hypertension förvärras när man tränar eller arbetar hårt.
F: Varför är pulmonell hypertension ett allvarligt tillstånd?
S: Pulmonell hypertension är ett allvarligt tillstånd eftersom det gör det svårare för hjärtat att pumpa blod och kan vara dödligt.
F: Vad är det fullständiga namnet på pulmonell hypertension?
S: Det fullständiga namnet på pulmonell hypertension är pulmonell arteriell hypertension, även om de flesta kallar det pah, ph eller pha.
F: Vad kan vissa mycket sjuka personer med pulmonell hypertension behöva för att överleva?
S: Vissa mycket sjuka personer med pulmonell hypertension kan behöva en lungtransplantation eller en hjärt-lungtransplantation för att överleva.
Relaterade artiklar
Författare
AlegsaOnline.com Pulmonell arteriell hypertension (PAH) Leandro Alegsa
URL: https://sv.alegsaonline.com/art/80022
Källor
- ncbi.nlm.nih.gov : PMID 8692238
- ncbi.nlm.nih.gov : PMID 14985486
- ncbi.nlm.nih.gov : PMID 10555089
- ncbi.nlm.nih.gov : PMID 13679525
- ncbi.nlm.nih.gov : PMID 10903931
- ncbi.nlm.nih.gov : PMID 14659797
- ncbi.nlm.nih.gov : PMID 15194171