Creutzfeldt–Jakobs sjukdom (CJD) – orsaker, symtom och prognos
Lär dig om Creutzfeldt–Jakobs sjukdom (CJD): orsaker, typiska symtom, snabbt förlopp och prognos. Fakta om prioner, diagnos och vård/support.
Creutzfeldt-Jakobs sjukdom (uttalas KROITS-felt YAH-kohb) eller CJD är en neurologisk sjukdom. Den är degenerativ (den blir värre med tiden), den kan inte botas och den leder alltid till döden. CJD kallas ibland för en mänsklig form av "galna ko-sjukan" (bovin spongiform encefalopati eller BSE). BSE är i själva verket en orsak till en sällsynt typ av Creutzfeldt-Jakobs sjukdom; de två är inte samma sjukdom.
CJD orsakas av ett smittämne som kallas prion. Prioner är proteiner som är felaktigt vikta. Prioner gör kopior av sig själva genom att ändra korrekt veckade proteiner till felveckade former. CJD leder till att hjärnvävnaden mycket snabbt blir ohälsosam. När sjukdomen förstör hjärnan utvecklas hål i hjärnan. Hjärnans konsistens förändras och blir som en kökssvamp.
Bildgalleri
6 Bilder




Olika former av CJD
Det finns flera former av CJD:
- Sporadisk CJD – den vanligaste formen. Uppstår utan känd orsak, oftast hos äldre vuxna.
- Familjär (ärftlig) CJD – orsakas av mutationer i genen för prionproteinet (PRNP) och kan drabba flera släktingar.
- Iatrogen CJD – mycket sällsynt, överförd genom medicinska procedurer (t.ex. kontaminerade kirurgiska instrument, dura-mater-transplantat eller hypofyshormon från humana källor).
- Variant CJD (vCJD) – kopplas till exponering för BSE-smittat nötkött. Denna form skiljer sig delvis i kliniskt förlopp och drabbar ofta yngre personer.
Symtom
Symtomen uppträder ofta snabbt och progredierar snabbt. Vanliga tecken är:
- Snabbt fortskridande demens – minnesstörning, förvirring och nedsatt omdöme.
- Myoklonier – plötsliga, ryckande muskelkramper (vanligt vid sporadisk CJD).
- Koordinationssvårigheter och ataksi (svårigheter att gå och hålla balansen).
- Synstörningar eller synbortfall.
- Behaviorella förändringar, depression eller apati.
- I senare skeden svårigheter att svälja, talstörningar och koma.
Det är viktigt att notera att CJD inte sprids genom vardaglig social kontakt som kramar eller pussar.
Diagnos
Diagnosen baseras på klinisk bild, neurologisk undersökning och kompletterande tester:
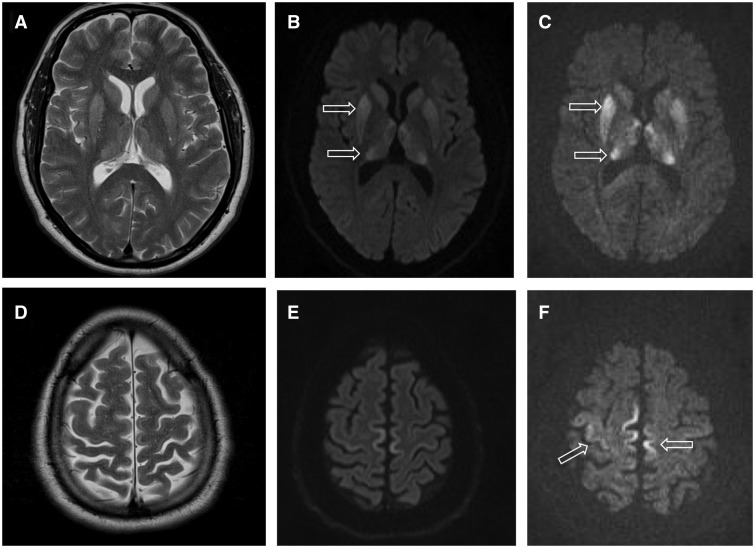
- MRT av hjärnan (DWI/FLAIR) visar ofta typiska förändringar i basala ganglier eller cortex ("cortical ribboning").
- EEG kan visa karakteristiska periodiska skarvkomplex hos vissa patienter.
- Likvoranalys (ryggmärgsvätska) för proteiner som 14-3-3, S100 eller neuron-specifika enolaser kan stödja diagnos, men är inte helt specifika.
- RT-QuIC (real-time quaking-induced conversion) är idag ett mycket känsligt och specifikt test som kan påvisa prionaktiva ämnen i likvor eller nässköljprov.
- Definitiv diagnos ställs vid hjärnbiopsi eller obduktion där spongiforma förändringar och onormalt prionprotein (PrPSc) kan påvisas.
Behandling och prognos
Det finns för närvarande ingen botande behandling för CJD. Vård är i huvudsak symptomatisk och palliativ och inriktas på att lindra symtom och ge stöd för patient och anhöriga. Exempel på åtgärder:
- Mediciner mot myoklonier (t.ex. levetiracetam eller valproat) och mot ångest eller sömnstörningar.
- Stöd för näring och vätska, fysioterapi och vård i livets slutskede.
Prognosen är oftast mycket allvarlig. Vid sporadisk CJD är sjukdomsförloppet vanligen snabbt — medianöverlevnad är ofta bara några månader efter symtomdebut och de flesta dör inom ett år. Familjär och variant CJD kan ha något annorlunda förlopp och variation i överlevnadstid.
Smittvägar och förebyggande
Prioner är ovanliga smittämnen: de är proteiner utan DNA eller RNA och är mycket motståndskraftiga mot vanliga sterilisationsmetoder. Viktiga punkter för att minska risk:
- Strikta infektionskontrollrutiner i sjukvården, särskilt vid hantering av hjärn- eller nervvävnad.
- Specifika sterilisationsmetoder (t.ex. NaOH och långvarig autoklavering under vissa förhållanden) för instrument som kan ha utsatts för prioner.
- Begränsningar vid blodgivning och noggrannhet kring transplantationer och vävnadsdonationer, särskilt i områden med känd risk för vCJD.
Förekomst och forskning
CJD är mycket sällsynt: den sporadiska formen förekommer i ungefär 1–2 fall per miljon invånare och år globalt. Forskning pågår för att bättre förstå prioners mekanismer, utveckla mer tillförlitliga diagnosmetoder tidigt i sjukdomsförloppet och pröva läkemedel som kan bromsa prionspridning eller skydda nervceller. Ett antal experimentella behandlingar och antikroppsstrategier utvärderas, men inga har visat sig vara botande i klinisk praxis än så länge.
Sammanfattning
Creutzfeldt–Jakobs sjukdom är en allvarlig, snabbt progredierande och dödlig prionsjukdom i centrala nervsystemet. Den kan uppstå sporadiskt, ärftligt, iatrogen eller som en variant relaterad till BSE. Diagnos bygger på klinik och särskilda undersökningar, och behandling är i nuläget stödjande. Förebyggande åtgärder i sjukvård och livsmedelsövervakning är viktiga för att minska överföringsrisker.
Typer och orsaker till CJD
Olika typer av CJD kan vara:
- variant (vCJD):
Denna typ av CJD kan orsakas av att man äter mat som innehåller prioner, som kött från kor som har BSE ("galna ko-sjukan"). Detta är dock en mycket ovanlig orsak till CJD.
- sporadisk (sCJD):
Detta är den vanligaste typen av CJD. 85 % av fallen av CJD är sporadisk CJD. Ingen vet vad som orsakar sCJD; det verkar ske slumpmässigt.
- familjär (fCJD):
De flesta av de övriga 15 % av fallen av CJD är familjära CJD-fall. Detta är en form av CJD som förekommer i familjer.
- iatrogen:
Denna form av CJD orsakas vanligtvis av ett medicinskt ingrepp där en person får blod eller vävnad från någon som har CJD. En person kan till exempel få iatrogen CJD om han eller hon får en blodtransfusion eller en hornhinnetransplantation från någon som har CJD.
Tecken och symtom
Det första symptomet på CJD är demens, som förvärras mycket snabbt.Demens orsakar minnesförlust, personlighetsförändringar och hallucinationer.
Andra vanliga psykiska symtom är:
Fysiska symtom på CJD inkluderar ofta:
- Svårigheter att tala
- Ryckiga rörelser (myoklonus)
- Problem med balansen (ataxi)
- Svårigheter att gå
- Skaka eller vara stel
- Problem med synen
- Sväljningssvårigheter, vilket kan göra det svårt eller omöjligt att äta.
- Problem med hosta, vilket kan orsaka lunginflammation
- Rörelser som patienten inte kan kontrollera (dyskinesi).
De flesta som drabbas av CJD dör inom sex månader efter att de första symptomen uppstod. Ofta dör de av lunginflammation orsakad av hosta. Ungefär 15 procent av patienterna överlever i två eller fler år. Vissa patienter har levt 4-5 år med mestadels mentala symtom tills sjukdomen förvärras och orsakar fler fysiska symtom. När detta sker dör personerna vanligtvis inom ett år.
Symptomen på CJD orsakas av att allt fler av hjärnans nervceller dör. När forskare tittar på hjärnvävnad från en CJD-patient i mikroskop kan de se många små hål där hela områden med nervceller har dött.
Diagnos
Läkare kan misstänka CJD när en person har vissa symtom. Till exempel förvärras demenssjukdomar vanligtvis långsamt. Demens som förvärras mycket snabbt är ovanligt. Tillsammans med symtom som ryckiga rörelser kan dessa symtom tyda på möjlig CJD.
Därefter kan tester göras för att visa om personen har CJD. Dessa tester omfattar följande:
- Elektroencefalografi (EEG): Detta test visar den elektriska aktiviteten i hjärnan. Läkaren kan ofta se förändringar i EEG som är vanliga hos personer med CJD. Vilken typ av förändringar som visas på EEG beror på vilken typ av CJD patienten har och hur långt sjukdomen har kommit.
- Lumbalpunktion (spinalpunktion): Detta test gör det möjligt att undersöka cerebrospinalvätskan (vätskan som omger hjärnan och ryggmärgen) och leta efter ett specifikt protein ("14-3-3-protein").
- MRT av hjärnan: Ett test som använder en mycket stark magnet för att ta bilder av hjärnan.
- Biopsi: För att göra en biopsi använder en kirurg en nål för att ta en liten bit vävnad från kroppen så att läkarna kan titta på den i ett mikroskop. vCJD kan diagnostiseras med en biopsi av tonsillerna. För alla andra typer av CJD är en biopsi av hjärnan det enda sättet att säkert avgöra om en person har CJD. Men eftersom en biopsi av hjärnan kan orsaka hjärnskador, görs en hjärnbiopsi vanligtvis inte om andra tester redan har visat att en person troligen har CJD.

Behandling
Det finns ännu inte någon behandling som botar CJD eller ens bromsar dess effekter. Många experiment görs för att försöka hitta behandlingar.
Idag är den enda behandlingen av CJD läkemedel som behandlar sjukdomens symtom och hjälper patienterna att må bättre. Patienter som har kramper kan till exempel få kramplösande läkemedel. Bensodiazepiner kan göra att muskelryckningar inträffar mindre ofta.
Patienterna kan också välja att genomgå medicinska ingrepp för att hjälpa till att lindra svåra symtom. CJD kan till exempel orsaka så stora problem med att svälja att en person inte kan äta. En del personer med CJD väljer att få en matningssond insatt när de inte längre kan äta. Detta är en slang som går in i magen, så att särskild vätska kan ges direkt in i magen för att ge personen näring.
Relaterade sidor
- Prion
- Prionsjukdom
- Terminalsjukdom
Frågor och svar
Fråga: Vad är Creutzfeldt-Jakobs sjukdom?
S: Creutzfeldt-Jakobs sjukdom (CJD) är en neurologisk sjukdom som är degenerativ, obotlig och alltid dödlig.
F: Finns det ett botemedel mot CJD?
S: Nej, det finns inget botemedel mot CJD.
F: Varför kallas CJD ibland för en mänsklig form av "galna kosjukan"?
S: CJD kallas ibland för en mänsklig form av "galna ko-sjukan" eftersom bovin spongiform encefalopati (BSE), som är en orsak till en sällsynt typ av CJD, är allmänt känd som "galna ko-sjukan".
F: Vad är orsaken till CJD?
S: CJD orsakas av ett smittämne som kallas prion, vilket är ett protein som är felaktigt veckat och som kan göra kopior av sig självt genom att ändra korrekt veckade proteiner till felaktigt veckade proteiner.
F: Vad händer med hjärnvävnaden vid CJD?
S: CJD leder till att hjärnvävnaden mycket snabbt blir ohälsosam, vilket resulterar i att det uppstår hål i hjärnan och att hjärnans konsistens förändras så att den blir som en kökssvamp.
F: Är BSE samma sjukdom som CJD?
S: Nej, BSE är inte samma sjukdom som CJD, utan är faktiskt en orsak till en sällsynt typ av CJD.
F: Hur orsakar prioner CJD?
S: Prioner orsakar CJD genom att de veckas felaktigt och gör kopior av sig själva på bekostnad av korrekt veckade proteiner i hjärnan. Detta leder till att frisk hjärnvävnad förstörs och att de hål som är karakteristiska för sjukdomen utvecklas.
Relaterade artiklar
Författare
AlegsaOnline.com Creutzfeldt–Jakobs sjukdom (CJD) – orsaker, symtom och prognos Leandro Alegsa
URL: https://sv.alegsaonline.com/art/24152
Källor
- cdc.gov : "CJD (Creutzfeldt–Jakob Disease, Classic)"
- ncbi.nlm.nih.gov : "Creutzfeldt–Jakob disease: Transmissible spongiform encephalopathy; vCJD; CJD; Jacob-Creutzfeldt disease"
- bmj.com : "Bovine spongiform encephalopathy and variant Creutzfeldt–Jakob disease"
- doi.org : 10.1101/SQB.1996.061.01.052
- pubmed.ncbi.nlm.nih.gov : 9246478
- www3.interscience.wiley.com : "MM2-cortical-type sporadic Creutzfeldt–Jakob disease with early stage cerebral cortical pathology presenting with a rapidly progressive clinical course"
- doi.org : 10.1111/j.1440-1789.2008.00904.x
- pubmed.ncbi.nlm.nih.gov : 18410280
- who.int : who.int: "Fact sheets no 180: Variant Creutzfeldt-Jakob disease" Feb 2012 ed.
- prion.ucl.ac.uk : "Sporadic Prion Disease"
- doi.org : 10.1056/NEJM198610163151605
- pubmed.ncbi.nlm.nih.gov : 3762620
- prion.ucl.ac.uk : "Sporadic Prion Disease"
- merckmanuals.com : "Creutzfeldt–Jakob Disease (CJD)"
- ncbi.nlm.nih.gov : "Creutzfeldt-Jakob disease: Updated diagnostic criteria, treatment algorithm, and the utility of brain biopsy"